11Q24.2-25 Micro-Rearrangements in Autism Spectrum Disorders: Relation to Brain Structures
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

5-Azacytidine Inhibits the Proliferation of Bladder Cancer Cells Via Reversal of the Aberrant Hypermethylation of the Hepacam Gene
ONCOLOGY REPORTS 35: 1375-1384, 2016 5-Azacytidine inhibits the proliferation of bladder cancer cells via reversal of the aberrant hypermethylation of the hepaCAM gene XIAORONG WANG1, E. CHEN1, XUE YANG1, YIN WANG1, ZHEN QUAN2, XIAOHOU WU2 and CHUNLI LUO1 1Key Laboratory of Diagnostic Medicine designated by the Chinese Ministry of Education, Chongqing Medical University; 2Department of Urology, The First Affiliated Hospital of Chongqing Medical University, Chongqing, P.R. China Received June 14, 2015; Accepted July 24, 2015 DOI: 10.3892/or.2015.4492 Abstract. Hepatocyte cell adhesion molecule (hepaCAM), a cells. In addition, AZAC inhibited the proliferation of bladder tumor-suppressor gene, is rarely expressed in bladder carci- cancer cells and arrested cells at the G0/G1 phase. The in vivo noma. However, little is known concerning the mechanisms of results showed that expression of DNMT3A/3B and hepaCAM low hepaCAM expression in bladder cancer. Abnormal hyper- as well as tumor growth of nude mice were markedly altered methylation in the promoter plays a crucial role in cancer by which corresponded with the in vitro results. Due to the ability silencing tumor-suppressor genes, which is catalyzed by DNA to reactivate expression of hepaCAM and inhibit growth of methyltransferases (DNMTs). In the present study, a total of bladder cancer cells, AZAC may represent an effective treat- 31 bladder cancer and 22 adjacent tissues were assessed by ment for bladder cancer. immunohistochemistry to detect DNMT3A/3B and hepaCAM expression. Methylation of hepaCAM was determined by Introduction methylation‑specific polymerase chain reaction (MSP). The mRNA and protein levels of DNMT3A/3B and hepaCAM Hepatocyte cell adhesion molecule (hepaCAM), also known were determined by RT-PCR and western blot analysis after as GliaCAM, is located on chromosome 11q24 and encodes a treatment with 5-azacytidine (AZAC). -

Hepacam Inhibits Cell Proliferation and Invasion in Prostate Cancer by Suppressing Nuclear Translocation of the Androgen Receptor Via Its Cytoplasmic Domain
MOLECULAR MEDICINE REPORTS 19: 2115-2124, 2019 HepaCAM inhibits cell proliferation and invasion in prostate cancer by suppressing nuclear translocation of the androgen receptor via its cytoplasmic domain QINGFU DENG1, LI LUO2, ZHEN QUAN1, NANJING LIU2, ZHONGBO DU1, WEI SUN1, CHUNLI LUO2 and XIAOHOU WU1 1Department of Urology, First Affiliated Hospital of Chongqing Medical University;2 Key Laboratory of Diagnostics Medicine Designated by The Ministry of Education, Chongqing Medical University, Chongqing 400042, P.R. China Received May 23, 2018; Accepted December 12, 2018 DOI: 10.3892/mmr.2019.9841 Abstract. Hepatocyte cell adhesion molecule (HepaCAM) is other industrialized countries (1). Therefore, it is necessary to a tumour suppressor. However, the mechanism of HepaCAM elucidate the underlying pathophysiological processes of PCa function in prostate cancer (PCa) remains unknown. In with the advent of global ageing. the present study, HepaCAM, androgen receptor (AR) Hepatocyte cell adhesion molecule (HepaCAM) was first and Ran were analysed in 46 PCa tissue samples using detected in the liver (2), and it was later identified as a member immunohistochemistry. Subsequently, the influence of of the immunoglobulin superfamily. Members of the immu- HepaCAM and its cytoplasmic domain on cell proliferation, noglobulin superfamily are primarily localized at the cell migration, and invasion, and associated proteins was examined membrane and are composed of three domains: Extracellular, using MTT, wound healing, Transwell and western blotting transmembrane and cytoplasmic (3-5). The cytoplasmic assays, respectively. Furthermore, nuclear translocation of domain is fundamental to its biological function (2). Recent AR and Ran was analysed using immunofluorescence and studies have indicated that HepaCAM is present at low levels Western blot assays. -

Bioinformatic Analysis of Next‑Generation Sequencing Data to Identify Dysregulated Genes in Fibroblasts of Idiopathic Pulmonary Fibrosis
INTERNATIONAL JOURNAL OF MOleCular meDICine 43: 1643-1656, 2019 Bioinformatic analysis of next‑generation sequencing data to identify dysregulated genes in fibroblasts of idiopathic pulmonary fibrosis CHAU‑CHYUN SHEU1-3, WEI‑AN CHANG1,2, MING‑JU TSAI1-3, SSU‑HUI LIAO1, INN‑WEN CHONG2,3 and PO-LIN KUO1 1Graduate Institute of Clinical Medicine, College of Medicine, Kaohsiung Medical University; 2Division of Pulmonary and Critical Care Medicine, Kaohsiung Medical University Hospital; 3Department of Internal Medicine, School of Medicine, College of Medicine, Kaohsiung Medical University, Kaohsiung 807, Taiwan, R.O.C. Received October 7, 2018; Accepted January 29, 2019 DOI: 10.3892/ijmm.2019.4086 Abstract. Idiopathic pulmonary fibrosis (IPF) is a lethal and miRNA expression data, combined with GEO verifica- fibrotic lung disease with an increasing global burden. It is tion, finally identified Homo sapiens (hsa)-miR-1254-INKA2 hypothesized that fibroblasts have a number of functions that and hsa-miR-766-3p-INKA2 as the potential miRNA-mRNA may affect the development and progression of IPF. However, interactions in IPF fibroblasts. In summary, the results the present understanding of cellular and molecular mecha- of the present study suggest that dysregulation of PAX8, nisms associated with fibroblasts in IPF remains limited. hsa-miR-1254-INKA2 and hsa-miR-766-3p-INKA2 may The present study aimed to identify the dysregulated genes promote the proliferation and survival of IPF fibroblasts. In in IPF fibroblasts, elucidate their functions and explore the functional analysis of the dysregulated genes, a marked potential microRNA (miRNA)‑mRNA interactions. mRNA association between fibroblasts and the ECM was identified. -

Identification of Tgfβ-Related Genes Regulated in Murine Osteoarthritis and Chondrocyte Hypertrophy by Comparison of Multiple Microarray Datasets
http://hdl.handle.net/1765/109466 Identification of TGFβ-related genes regulated in murine osteoarthritis and chondrocyte hypertrophy by comparison of multiple microarray datasets Laurie M.G. de Kroon1,2,&, Guus G.H. van den Akker1,&, Bent Brachvogel3,4, Roberto Narcisi2, Daniele Belluoccio5, Florien Jenner6, John F. Bateman5, Christopher B. Little7, Pieter Brama8, Esmeralda N. Blaney Davidson1, Peter M. van der Kraan1, Gerjo J.V.M. van Osch2,9* 1Department of Rheumatology, Experimental Rheumatology, Radboud University Medical Center, Nijmegen, the Netherlands 2Department of Orthopedics, Erasmus MC University Medical Center, Rotterdam, the Netherlands 3Center for Biochemistry, Medical Faculty, University of Cologne, Cologne, Germany 4Department of Pediatrics and Adolescent Medicine, Experimental Neonatology, Medical Faculty, University of Cologne, Cologne, Germany 5Murdoch Childrens Research Institute, Royal Children’s Hospital, Parkville, Victoria, Australia 6Equine University Hospital, University of Veterinary Medicine, Vienna, Austria 7Raymond Purves Bone and Joint Research Laboratories, Kolling Institute of Medical Research, University of Sydney, St Leonards, New South Wales, Australia 8Veterinary Clinical Sciences, School of Veterinary Medicine, University College Dublin, Dublin, Ireland 9Department of Otorhinolaryngology, Erasmus MC University Medical Center, Rotterdam, the Netherlands &Both authors contributed equally *Corresponding author: Gerjo van Osch ([email protected]) Address for correspondence: Erasmus MC, Departments -

Breakpoint Mapping and Haplotype Analysis of Translocation T(1;12)(Q43;Q21.1) in Two Apparently Independent Families with Vascular Phenotypes
Received: 7 August 2017 | Revised: 9 October 2017 | Accepted: 11 October 2017 DOI: 10.1002/mgg3.346 ORIGINAL ARTICLE Breakpoint mapping and haplotype analysis of translocation t(1;12)(q43;q21.1) in two apparently independent families with vascular phenotypes Tiia Maria Luukkonen1,2 | Mana M. Mehrjouy3 | Minna Poyh€ onen€ 4,5 | Anna-Kaisa Anttonen6 | Paivi€ Lahermo1 | Pekka Ellonen1 | Lars Paulin7 | Niels Tommerup3 | Aarno Palotie1,8 | Teppo Varilo2,5 1Institute for molecular medicine Finland FIMM, University of Helsinki, Helsinki, Abstract Finland Background: The risk of serious congenital anomaly for de novo balanced 2Department of Health, National Institute translocations is estimated to be at least 6%. We identified two apparently inde- for Health and Welfare, Helsinki, Finland pendent families with a balanced t(1;12)(q43;q21.1) as an outcome of a “System- 3Wilhelm Johannsen Centre for atic Survey of Balanced Chromosomal Rearrangements in Finns.” In the first Functional Genome Research, Department of Cellular and Molecular family, carriers (n = 6) manifest with learning problems in childhood, and later Medicine, University of Copenhagen, with unexplained neurological symptoms (chronic headache, balance problems, Copenhagen, Denmark tremor, fatigue) and cerebral infarctions in their 50s. In the second family, two 4Clinical Genetics, Helsinki University Hospital, University of Helsinki, carriers suffer from tetralogy of Fallot, one from transient ischemic attack and one Helsinki, Finland from migraine. The translocation cosegregates with these vascular phenotypes and 5Department of Medical Genetics, neurological symptoms. University of Helsinki, Helsinki, Finland Methods and Results: We narrowed down the breakpoint regions using mate 6Laboratory of Genetics, HUSLAB, Helsinki, Finland pair sequencing. We observed conserved haplotypes around the breakpoints, 7Institute of Biotechnology, University of pointing out that this translocation has arisen only once. -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in -

Molecular Signatures Differentiate Immune States in Type 1 Diabetes Families
Page 1 of 65 Diabetes Molecular signatures differentiate immune states in Type 1 diabetes families Yi-Guang Chen1, Susanne M. Cabrera1, Shuang Jia1, Mary L. Kaldunski1, Joanna Kramer1, Sami Cheong2, Rhonda Geoffrey1, Mark F. Roethle1, Jeffrey E. Woodliff3, Carla J. Greenbaum4, Xujing Wang5, and Martin J. Hessner1 1The Max McGee National Research Center for Juvenile Diabetes, Children's Research Institute of Children's Hospital of Wisconsin, and Department of Pediatrics at the Medical College of Wisconsin Milwaukee, WI 53226, USA. 2The Department of Mathematical Sciences, University of Wisconsin-Milwaukee, Milwaukee, WI 53211, USA. 3Flow Cytometry & Cell Separation Facility, Bindley Bioscience Center, Purdue University, West Lafayette, IN 47907, USA. 4Diabetes Research Program, Benaroya Research Institute, Seattle, WA, 98101, USA. 5Systems Biology Center, the National Heart, Lung, and Blood Institute, the National Institutes of Health, Bethesda, MD 20824, USA. Corresponding author: Martin J. Hessner, Ph.D., The Department of Pediatrics, The Medical College of Wisconsin, Milwaukee, WI 53226, USA Tel: 011-1-414-955-4496; Fax: 011-1-414-955-6663; E-mail: [email protected]. Running title: Innate Inflammation in T1D Families Word count: 3999 Number of Tables: 1 Number of Figures: 7 1 For Peer Review Only Diabetes Publish Ahead of Print, published online April 23, 2014 Diabetes Page 2 of 65 ABSTRACT Mechanisms associated with Type 1 diabetes (T1D) development remain incompletely defined. Employing a sensitive array-based bioassay where patient plasma is used to induce transcriptional responses in healthy leukocytes, we previously reported disease-specific, partially IL-1 dependent, signatures associated with pre and recent onset (RO) T1D relative to unrelated healthy controls (uHC). -

Identifying Genetic Interactions Associated with Late-Onset
1 Identifying Genetic Interactions Associated with Late-Onset 2 Alzheimer’s Disease 3 Charalampos S. Floudas, M.D, Ph.D. 1§ , Nara Um, M.D, M.S. 1, M. Ilyas Kamboh, Ph.D. 2, 4 Michael M. Barmada, Ph.D. 2, Shyam Visweswaran, M.D, Ph.D. 1,3 5 6 1Department of Biomedical Informatics, University of Pittsburgh, Pittsburgh, PA, USA s t 7 2Department of Human Genetics, University of Pittsburgh, Pittsburgh, PA, USA n i r 8 3The Intelligent Systems Program, University of Pittsburgh, Pittsburgh, PA, USA P e r 9 P 10 §Corresponding author 11 12 13 14 15 16 17 Email addresses : 18 CSF: [email protected] 19 SV: [email protected] 20 1 PeerJ PrePrints | https://peerj.com/preprints/123v2/ | v2 received: 11 Dec 2013, published: 11 Dec 2013, doi: 10.7287/peerj.preprints.123v2 21 Abstract 22 Background 23 Identifying genetic interactions in data obtained from genome-wide association studies (GWASs) 24 can help in understanding the genetic basis of complex diseases. The large number of single 25 nucleotide polymorphisms (SNPs) in GWASs however makes the identification of genetic 26 interactions computationally challenging. We developed the Bayesian Combinatorial Method s t n 27 (BCM) that can identify pairs of SNPs that in combination have high statistical association with i r P 28 disease. e r 29 Results P 30 We applied BCM to two late-onset Alzheimer’s disease (LOAD) GWAS datasets to identify 31 SNP-SNP interactions between a set of known SNP associations and the dataset SNPs. For 32 evaluation we compared our results with those from logistic regression, as implemented in 33 PLINK. -

Depression-Associated Gene Negr1-Fgfr2 Pathway Is Altered by Antidepressant Treatment
cells Article Depression-Associated Gene Negr1-Fgfr2 Pathway Is Altered by Antidepressant Treatment Lucia Carboni 1,* , Francesca Pischedda 2, Giovanni Piccoli 2, Mario Lauria 3,4 , Laura Musazzi 5 , Maurizio Popoli 6, Aleksander A. Mathé 7 and Enrico Domenici 2,4 1 Department of Pharmacy and Biotechnology, Alma Mater Studiorum Università di Bologna, 40126 Bologna, Italy 2 Department of Cellular, Computational and Integrative Biology, University of Trento, 38123 Trento, Italy; [email protected] (F.P.); [email protected] (G.P.); [email protected] (E.D.) 3 Department of Mathematics, University of Trento, 38123 Trento, Italy; [email protected] 4 Fondazione The Microsoft Research—University of Trento Centre for Computational and Systems Biology (COSBI), 38068 Rovereto (Trento), Italy 5 School of Medicine and Surgery, University of Milano-Bicocca, 20900 Monza, Italy; [email protected] 6 Laboratory of Neuropsychopharmacology and Functional Neurogenomics, Dipartimento di Scienze Farmaceutiche, Università degli Studi di Milano, 20133 Milano, Italy; [email protected] 7 Karolinska Institutet, Department of Clinical Neuroscience, SE-11221 Stockholm, Sweden; [email protected] * Correspondence: [email protected]; Tel.: +39-051-209-1793 Received: 30 June 2020; Accepted: 28 July 2020; Published: 31 July 2020 Abstract: The Negr1 gene has been significantly associated with major depression in genetic studies. Negr1 encodes for a cell adhesion molecule cleaved by the protease Adam10, thus activating Fgfr2 and promoting neuronal spine plasticity. We investigated whether antidepressants modulate the expression of genes belonging to Negr1-Fgfr2 pathway in Flinders sensitive line (FSL) rats, in a corticosterone-treated mouse model of depression, and in mouse primary neurons. -

3,5-T3 in Brain and Liver Uncovers Novel Roles for Thyroid
www.nature.com/scientificreports OPEN Diferential transcriptome regulation by 3,5-T2 and 3′,3,5-T3 in brain and liver uncovers novel Received: 25 May 2017 Accepted: 16 October 2017 roles for thyroid hormones in tilapia Published: xx xx xxxx A. Olvera1, C. J. Martyniuk2, N. Buisine3, V. Jiménez-Jacinto4, A. Sanchez-Flores4, L. M. Sachs3 & A. Orozco 1 Although 3,5,3′-triiodothyronine (T3) is considered to be the primary bioactive thyroid hormone (TH) due to its high afnity for TH nuclear receptors (TRs), new data suggest that 3,5-diiodothyronine (T2) can also regulate transcriptional networks. To determine the functional relevance of these bioactive THs, RNA-seq analysis was conducted in the cerebellum, thalamus-pituitary and liver of tilapia treated with equimolar doses of T2 or T3. We identifed a total of 169, 154 and 2863 genes that were TH- responsive (FDR < 0.05) in the tilapia cerebellum, thalamus-pituitary and liver, respectively. Among these, 130, 96 and 349 genes were uniquely regulated by T3, whereas 22, 40 and 929 were exclusively regulated by T2 under our experimental paradigm. The expression profles in response to TH treatment were tissue-specifc, and the diversity of regulated genes also resulted in a variety of diferent pathways being afected by T2 and T3. T2 regulated gene networks associated with cell signalling and transcriptional pathways, while T3 regulated pathways related to cell signalling, the immune system, and lipid metabolism. Overall, the present work highlights the relevance of T2 as a key bioactive hormone, and reveals some of the diferent functional strategies that underpin TH pleiotropy. -
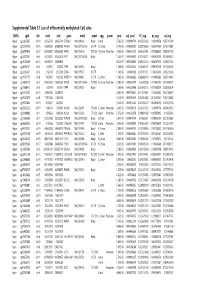
Supplementary Table S1 Kim Et Al.Xlsx
Supplemental Table S1. List of differentially methylated CpG sites DMRs cgid chr start end gene nmid annot cpg prom pval adj. pval FC_avg nc_avg cd_avg Hyper cg12827637 chr14 69256790 69256791 ZFP36L1 NM_004926 Body Island . 5.76E-05 0.000989714 4.465355492 0.06419878 0.287016909 Hyper cg02072495 chr15 60689284 60689285 ANXA2 NM_001136015 5'UTR N_Shore . 2.04E-05 0.000560282 3.808196653 0.056572464 0.215719887 Hyper cg26147845 chr12 132433837 132433838 EP400 NM_015409 TSS1500N_Shore Promoter 3.92E-06 0.000301217 3.643351981 0.077868629 0.283967159 Hyper cg23737061 chr3 60226327 60226328 FHIT NM_001166243 Body . 1.65E-07 0.000239667 3.616168212 0.093920309 0.339893251 Hyper cg18215449 chr12 66089472 66089473 . 4.22E-07 0.000239667 3.565225234 0.064067475 0.228671502 Hyper cg24082121 chr5 672871 672872 TPPP NM_007030 Body . 1.78E-05 0.000525064 3.505843071 0.089095718 0.312606189 Hyper cg23032421 chr3 3152037 3152038 IL5RA NM_175727 5'UTR . 1.04E-05 0.00041338 3.371418731 0.114213604 0.385299026 Hyper cg11737172 chr8 1923051 1923052 KBTBD11 NM_014867 5'UTR S_Shore . 1.38E-05 0.000466865 3.368483202 0.108495063 0.365700645 Hyper cg16886175 chr7 114562062 114562063 MDFIC NM_001166345 TSS200 N_Shore Promoter 7.28E-07 0.000243374 3.35638256 0.101463003 0.340784291 Hyper cg10986412 chr5 672909 672910 TPPP NM_007030 Body . 1.56E-06 0.000255546 3.316242102 0.071981379 0.238939304 Hyper cg02025737 chr15 33384750 33384751 . 3.80E-05 0.000776402 3.312390381 0.13643852 0.452168879 Hyper cg06243675 chr8 11801364 11801365 . 5.23E-05 0.000932639 3.254014485 0.016251643 0.053108482 Hyper cg09577804 chr10 3235531 3235532 . 5.04E-05 0.000912242 3.247042327 0.043496295 0.141459016 Hyper cg23057220 chr19 1356314 1356315 MUM1 NR_024247 TSS200 S_Shore Promoter 3.65E-05 0.000758178 3.204370027 0.14549078 0.466426732 Hyper cg06984883 chr1 1243563 1243564 PUSL1 NM_153339 TSS1500Island Promoter 5.07E-06 0.000323878 3.198831858 0.005047892 0.01636724 Hyper cg15963463 chr1 25253236 25253237 RUNX3 NM_001031680 Body N_Shelf . -

Genome-Wide Homozygosity Patterns and Evidence for Selection in a Set of European and Near Eastern Horse Breeds
Article Genome-Wide Homozygosity Patterns and Evidence for Selection in a Set of European and Near Eastern Horse Breeds Gertrud Grilz-Seger 1,*, Markus Neuditschko 2, Anne Ricard 3, Brandon Velie 4,5, Gabriella Lindgren 4,6,, Matjaz Mesarič 7, Marko Cotman 8, Michaela Horna 9, Max Dobretsberger 1, Gottfried Brem 1 and Thomas Druml 1 1 Institute of Animal Breeding and Genetics, University of Veterinary Sciences Vienna, Veterinärplatz 1, 1210 Vienna, Austria; [email protected] (M.D.); [email protected] (G.B.); [email protected] (T.D.) 2 Agroscope, Swiss National Stud Farm, Les Longs Prés, CH-1580 Avenches, Switzerland; [email protected] 3 UMR 1313 Génétique Animale et Biologie Intégrative, Institut National de la Recherche Agronomique, Domaine de Vilvert, Bat 211, 78352 Jouy-en-Josas, France; [email protected] 4 Department of Animal Breeding & Genetics, Swedish University of Agricultural Sciences, Ulls väg 26, 750 07 Uppsala, Sweden; [email protected] (B.V.); [email protected] (G.L.) 5 School of Life and Environmental Sciences, University of Sydney, Eastern Ave, 2006 NSW Sydney, Australia 6 Livestock Genetics, Department of Biosystems, KU Leuven, 3001 Leuven, Belgium 7 Clinic for Reproduction and Large Animals, University of Ljubljana, Veterinary, Faculty, Cesta v Mestni log 47, 1000 Ljubljana, Slovenia; [email protected] 8 Institute for Preclinical Sciences, University of Ljubljana, Veterinary Faculty, Gerbičeva 60, 1000 Ljubljana, Slovenia; [email protected] 9 Department of Animal Husbandry, Slovak University of Agriculture in Nitra, Tr. A. Hlinku 2, 949 76 Nitra, Slovakia; [email protected] * Correspondence: [email protected] Received: 14 May 2019; Accepted: 26 June 2019; Published: 28 June 2019 Abstract: Intensive artificial and natural selection have shaped substantial variation among European horse breeds.