Purification and Characterization of P-Type Atpases a Major Qualifying Report Worcester Polytechnic Institute Degree of Bachelo
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Cellular Transport Notes About Cell Membranes
Cellular Transport Notes @ 2011 Center for Pre-College Programs, New Jersey Institute of Technology, Newark, New Jersey About Cell Membranes • All cells have a cell membrane • Functions: – Controls what enters and exits the cell to maintain an internal balance called homeostasis TEM picture of a – Provides protection and real cell membrane. support for the cell @ 2011 Center for Pre-College Programs, New Jersey Institute of Technology, Newark, New Jersey 1 About Cell Membranes (continued) 1.Structure of cell membrane Lipid Bilayer -2 layers of phospholipids • Phosphate head is polar (water loving) Phospholipid • Fatty acid tails non-polar (water fearing) • Proteins embedded in membrane Lipid Bilayer @ 2011 Center for Pre-College Programs, New Jersey Institute of Technology, Newark, New Jersey Polar heads Fluid Mosaic love water Model of the & dissolve. cell membrane Non-polar tails hide from water. Carbohydrate cell markers Proteins @ 2011 Center for Pre-College Programs, New Jersey Institute of Technology, Newark, New Jersey 2 About Cell Membranes (continued) • 4. Cell membranes have pores (holes) in it • Selectively permeable: Allows some molecules in and keeps other molecules out • The structure helps it be selective! Pores @ 2011 Center for Pre-College Programs, New Jersey Institute of Technology, Newark, New Jersey Structure of the Cell Membrane Outside of cell Carbohydrate Proteins chains Lipid Bilayer Transport Protein Phospholipids Inside of cell (cytoplasm) @ 2011 Center for Pre-College Programs, New Jersey Institute of Technology, Newark, New Jersey 3 Types of Cellular Transport • Passive Transport celldoesn’tuseenergy 1. Diffusion 2. Facilitated Diffusion 3. Osmosis • Active Transport cell does use energy 1. -

Passive and Active Transport
Passive and Active Transport 1. Thermodynamics of transport 2. Passive-mediated transport 3. Active transport neuron, membrane potential, ion transport Membranes • Provide barrier function – Extracellular – Organelles • Barrier can be overcome by „transport proteins“ – To mediate transmembrane movements of ions, Na+, K+ – Nutrients, glucose, amino acids etc. – Water (aquaporins) 1) Thermodynamics of Transport • Aout <-> Ain (ressembles a chemical equilibration) o‘ • GA - G A = RT ln [A] • ∆GA = GA(in) - GA(out) = RT ln ([A]in/[A]out) • GA: chemical potential of A o‘ • G A: chemical potential of standard state of A • If membrane has a potential, i.e., plasma membrane: -100mV (inside negative) then GA is termed the electrochemical potential of A Two types of transport across a membrane: o Nonmediated transport occurs by passive diffusion, i.e., O2, CO2 driven by chemical potential gradient, i.e. cannot occur against a concentration gradient o Mediated transport occurs by dedicated transport proteins 1. Passive-mediated transport/facilitated diffusion: [high] -> [low] 2. Active transport: [low] -> [high] May require energy in form of ATP or in form of a membrane potential 2) Passive-mediated transport Substances that are too large or too polar to diffuse across the bilayer must be transported by proteins: carriers, permeases, channels and transporters A) Ionophores B) Porins C) Ion Channels D) Aquaporins E) Transport Proteins A) Ionophores Organic molecules of divers types, often of bacterial origin => Increase the permeability of a target membrane for ions, frequently antibiotic, result in collapse of target membrane potential by ion equilibration 1. Carrier Ionophore, make ion soluble in membrane, i.e. valinomycin, 104 K+/sec 2. -

The Electrochemical Gradient of Protons and Its Relationship to Active Transport in Escherichia Coli Membrane Vesicles
Proc. Natl. Acad. Sci. USA Vol. 73, No. 6, pp. 1892-1896, June 1976 Biochemistry The electrochemical gradient of protons and its relationship to active transport in Escherichia coli membrane vesicles (flow dialysis/membrane potential/energy transduction/lipophilic cations/weak acids) SOFIA RAMOS, SHIMON SCHULDINER*, AND H. RONALD KABACK The Roche Institute of Molecular Biology, Nutley, New Jersey 07110 Communicated by B. L. Horecker, March 17, 1976 ABSTRACT Membrane vesicles isolated from E. coli gen- presence of valinomycin), a respiration-dependent membrane erate a trans-membrane proton gradient of 2 pH units under potential (AI, interior negative) of approximately -75 mV in appropriate conditions when assayed by flow dialysis. Using E. coli membrane vesicles has been documented (6, 13, 14). the distribution of weak acids to measure the proton gradient (ApH) and the distribution of the lipophilic cation triphenyl- Moreover it has been shown that the potential causes the ap- methylphosphonium to measure the electrical potential across pearance of high affinity binding sites for dansyl- and azido- the membrane (AI), the vesicles are shown to generate an phenylgalactosides on the outer surface of the membrane (4, electrochemical proton gradient (AiH+) of approximately -180 15) and that the potential is partially dissipated as a result of mV at pH 5.5 in the presence of ascorbate and phenazine lactose accumulation (6). Although these findings provide ev- methosulfate, the major component of which is a ApH of about idence for the chemiosmotic hypothesis, it has also been dem- -110 mV. As external pH is increased, ApH decreases, reaching o at pH 7.5 and above, while AI remains at about -75 mV and onstrated (6, 16) that vesicles are able to accumulate lactose and internal pH remains at pH 7.5. -

Specific Copb Transporter: Revising P1B-Type Atpase Classification
+ Cu -specific CopB transporter: Revising P1B-type ATPase classification Rahul Purohita,b, Matthew O. Rossa,b, Sharon Bateluc, April Kusowskic,d, Timothy L. Stemmlerc,d, Brian M. Hoffmana,b, and Amy C. Rosenzweiga,b,1 aDepartment of Molecular Biosciences, Northwestern University, Evanston, IL 60208; bDepartment of Chemistry, Northwestern University, Evanston, IL 60208; cDepartment of Pharmaceutical Sciences, Wayne State University, Detroit, MI 48201; and dSchool of Medicine, Wayne State University, Detroit, MI 48201 Contributed by Amy C. Rosenzweig, January 12, 2018 (sent for review December 14, 2017; reviewed by Megan M. McEvoy and Gabriele Meloni) The copper-transporting P1B-ATPases, which play a key role in cellu- metal specificities of the P1B-5 (PCP motif), P1B-6 (SCA motif), lar copper homeostasis, have been divided traditionally into two and P1B-7-ATPases (CSC motif) remain unclear, although some 2+ 2+ subfamilies, the P1B-1-ATPases or CopAs and the P1B-3-ATPases or evidence links the P1B-5-ATPases to Ni and Fe (20, 21). The + CopBs. CopAs selectively export Cu whereas previous studies and remaining two groups are the copper transporters. The P1B-1- + bioinformatic analyses have suggested that CopBs are specific for ATPases, which include ATP7A and ATP7B, transport Cu 2+ 2+ Cu export. Biochemical and spectroscopic characterization of (9, 22), whereas the P1B-3-ATPases are proposed to transport Cu Sphaerobacter thermophilus CopB (StCopB) show that, while it does (23, 24). These two subfamilies differ from one another in several 2+ bind Cu , the binding site is not the prototypical P1B-ATPase trans- ways. First, the TM helix 4 motif is CPC in the P1B-1-ATPases and membrane site and does not involve sulfur coordination as proposed CPH in the P1B-3-ATPases. -

Biology Passive & Active Transport April 30, 2020
High School Science Virtual Learning Biology Passive & Active Transport April 30, 2020 High School General Biology Lesson: Passive & Active Transport Objective/Learning Target: Students will understand how passive and active transports work. Bell Ringer Activity 1. If someone is being active what does that mean? 2. If someone is being passive what does that mean? Bell Ringer Answers 1. If someone is being active that means they are marked by energetic activity. 2. If someone is being passive they are accepting what happens to others without an active response. Keep these definitions in mind as we discuss the differences between what active and passive transport are in biology. Let’s Get Started! Lesson Activity: Directions: 1. Watch this video. 2. Create a Venn Diagram like the one you see here ---> 3. Compare and contrast Active and Passive Transport by the information you learn from the video. Lesson Questions Answers Venn Diagram Examples: Practice Questions 1. What is passive transport? 2. What is active transport? 3. What is the difference between diffusion and osmosis? 4. What is the difference between endocytosis and exocytosis? 5. What is the differences between facilitated diffusion and active transport by a protein pump? Answers to Practice Questions 1. Passive transport is the movement of materials across the cell membrane without using cellular energy. 2. Active transport is the movement of materials against a concentration difference; it requires energy. 3. In diffusion, both solvent and solute particles are free to move; however, in osmosis only water molecules cross the semipermeable membrane. Answers to Practice Questions Continued 4. -
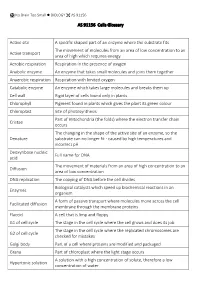
AS 91156 Cells Glossary Active Site a Specific Shaped Part of an Enzyme Where the Substrate Fits Active Transport the Movement
No Brain Too Small BIOLOGY AS 91156 AS 91156 Cells Glossary Active site A specific shaped part of an enzyme where the substrate fits The movement of molecules from an area of low concentration to an Active transport area of high which requires energy Aerobic respiration Respiration in the presence of oxygen Anabolic enzyme An enzyme that takes small molecules and joins them together Anaerobic respiration Respiration with limited oxygen Catabolic enzyme An enzyme which takes large molecules and breaks them up Cell wall Rigid layer of cells found only in plants Chlorophyll Pigment found in plants which gives the plant its green colour Chloroplast Site of photosynthesis Part of mitochondria (the folds) where the electron transfer chain Cristae occurs The changing in the shape of the active site of an enzyme, so the Denature substrate can no longer fit - caused by high temperatures and incorrect pH Deoxyribose nucleic Full name for DNA acid The movement of materials from an area of high concentration to an Diffusion area of low concentration DNA replication The copying of DNA before the cell divides Biological catalysts which speed up biochemical reactions in an Enzymes organism A form of passive transport where molecules move across the cell Facilitated diffusion membrane through the membrane proteins Flaccid A cell that is limp and floppy G1 of cell cycle The stage in the cell cycle where the cell grows and does its job The stage in the cell cycle where the replicated chromosomes are G2 of cell cycle checked for mistakes Golgi body Part -

Living Environment Vocabulary by Prentice Hall 2001 Review Book Unit
Living Environment Vocabulary By Prentice Hall 2001 Review Book Unit Similarities and Topic 1 Differences Among Living Organisms cell the basic unit of structure and function that makes up all organisms metabolism all the chemical reactions that occur within the cells of an organism homeostasis the ability of an organism to maintain a stable internal environment even when the external environment changes reproduction the process by which organisms produce new organisms of the same type cell respiration the process in which nutrients are broken apart, releasing the chemical energy stored in them synthesis a life process that involves combining simple substances into more complex substances organic term used to describe molecules that contain both hydrogen and carbon inorganic a type of molecule that does not contain both carbon and hydrogen but can contain any other combination of elements organelle a structure within the cell that carries out a specific function tissues a group of specialized cells that perform a specific function organ a body structure made of different kinds of tissues combined to perform a specific function organ system several organs that work together to perform a major function in the body cytoplasm the jellylike substance that is between the cell membrane and the nucleus and that contains specialized structures nucleus a large structure within a cell that controls the cell’s metabolism and stores genetic information, including chromosomes and DNA vacuoles storage sacs within the cytoplasm of a cell that may contain -

Conformational Model of Active Transport John H
Proceedings of the National Academy of Sciences Vol. 67, No. 2, pp. 550-559, October 1970 Conformational Model of Active Transport John H. Young*, George A. Blondin, G. Vanderkooi, and D. E. Green INSTITUTE FOR ENZYME RESEARCH AND THEORETICAL CHEMISTRY INSTITUTE UNIVERSITY OF WISCONSIN, MADISON 53706 Read as an Invited Paper on the Conformational Basis of an Energy Transduction, April 29, 1970 Abstract. A model of active transport of monovalent cations in mitochondria is developed. The model is based on the coupling of electron transfer to the generation of a metastable protein conformation which in turn leads to the generation of an asymmetric surface charge, a membrane potential, and a redis- tribution of diffusible ions across the inner mitochondrial membrane. The ions at all times move spontaneously down an electrochemical potential gradient in this model so that there is no need to invoke the concept of an ion pump. It is shown that a wide variety of experimental facts can be rationalized in terms of the present model. In seeking an explanation for active transport, two mutually exclusive alterna- tives present themselves-either the ions move spontaneously down an electro- chemical potential gradient or they do not. Whereas many investigators in the field of active transport believe that ions are pumped against an electro- chemical potential gradient, the model we will present involves no ion pumps but only the spontaneous movement of ions down an electrochemical potential gradient. We will consider only one active transport system, the mitochondrion, and we will consider only monovalent cation translocation in the mitochondrion. Even for this special case we cannot yet claim to have a complete explanation of active transport. -

BBA 45 232 the EFFECT of METABOLIC INHIBITORS on TRANSPORT and EXCHANGE of AMINO ACIDS in EHRLICH ASCITES CELLS I. Cyanide and 2
128 BIOC/-IIMICA ET BIOPI-IYSICA ACTA BBA 45 232 THE EFFECT OF METABOLIC INHIBITORS ON TRANSPORT AND EXCHANGE OF AMINO ACIDS IN EHRLICH ASCITES CELLS JOHN A. JACQUEZ AND JAMES H. SHERMAN Departments of Physiology and Biostatistics, and Department of Physiology, The University of Michigan, Ann Arbor, Mich. (U.S.A.) (Received February 23rd, 1965) SUMMARY I. Cyanide and 2-deoxyglucose inhibit transport fluxes but do not inhibit exchange fluxes of amino acids in Ehrlich ascites cells. This is true whether the exchange flux is of the same order of magnitude as the transport flux or whether it is much greater. 2. These findings are compatible with the assumption that the linkage between the carrier system and energy metabolism is such as to either (i) increase the rate of outward movement of free carrier across the cell membrane, or (ii) increase the rate of dissociation of carrier-amino acid complex at the inner surface of the cell mem- brane. These findings are not compatible with the assumptions that the linkage is such as (iii) to increase the rate of movement of carrier-amino acid complex across the cell membrane, or (iv) to increase the rate of formation of the carrier-amino acid complex at the outer surface of the cell membrane. INTRODUCTION USS1NG1 used the term exchange diffusion to describe a process in which an ion exchanges across a membrane separating two phases by a mechanism which involves the formation of a complex with a constituent of the membrane which he called a carrier. He defined this as a one-for-one exchange of the same ion and there- fore considered it to be independent of any energy requirement. -

Description of Active Transport
Bioenergetics: Open Access Editorial Description of Active Transport Xinkun Wang * Higuchi Biosciences Center, University of Kansas, USA INTRODUCTION Active transport is the movement of molecules across a cell Turn on a fan and place it near the bag of popcorn. Inquire of membrane across a concentration gradient from a region of your students how this might alter the flow of air molecules. lower concentration to a region of higher concentration. To Active transport necessitates the use of a specific carrier molecule, achieve this movement, active transport necessitates the use of a protein, as well as the expenditure of cellular energy; as a result, cellular energy. The technique of transporting molecules across transfer across membranes can take place against a concentration a cellular membrane using cellular energy is known as active gradient. Substances migrate against the concentration gradient transport. Active transport transports molecules from a lower during active transport, from a low- concentration location to a concentration to a higher concentration, whereas passive high-concentration one. This is a "active" process because it transport can only move molecules from a higher concentration necessitates the application of energy (usually in the form of to a lower concentration. ive transport necessitates the use of ATP). Passive transportation is the polar opposite of active cellular energy to accomplish this movement. A protein pump transportation. Active transport is a methodof transport that uses energy in the form of ATP to move molecules from a low- makes use of the energy produced by respiration. Diffusion and concentration location to a high- concentration location during osmosis are passive processes that do not require the cell to active transport. -

5.3 Active Transport
Chapter 5 | Structure and Function of Plasma Membranes 159 fish actively take in salt through their gills and excrete diluted urine to rid themselves of excess water. Saltwater fish live in the reverse environment, which is hypertonic to their cells, and they secrete salt through their gills and excrete highly concentrated urine. In vertebrates, the kidneys regulate the water amount in the body. Osmoreceptors are specialized cells in the brain that monitor solute concentration in the blood. If the solute levels increase beyond a certain range, a hormone releases that slows water loss through the kidney and dilutes the blood to safer levels. Animals also have high albumin concentrations, which the liver produces, in their blood. This protein is too large to pass easily through plasma membranes and is a major factor in controlling the osmotic pressures applied to tissues. 5.3 | Active Transport By the end of this section, you will be able to do the following: • Understand how electrochemical gradients affect ions • Distinguish between primary active transport and secondary active transport Active transport mechanisms require the cell’s energy, usually in the form of adenosine triphosphate (ATP). If a substance must move into the cell against its concentration gradient—that is, if the substance's concentration inside the cell is greater than its concentration in the extracellular fluid (and vice versa)—the cell must use energy to move the substance. Some active transport mechanisms move small-molecular weight materials, such as ions, through the membrane. Other mechanisms transport much larger molecules. Electrochemical Gradient We have discussed simple concentration gradients—a substance's differential concentrations across a space or a membrane—but in living systems, gradients are more complex. -

Glossary.Pdf
Glossary Pronunciation Key accessory fruit A fruit, or assemblage of fruits, adaptation Inherited characteristic of an organ- Pronounce in which the fleshy parts are derived largely or ism that enhances its survival and reproduc- a- as in ace entirely from tissues other than the ovary. tion in a specific environment. – Glossary Ј Ј a/ah ash acclimatization (uh-klı¯ -muh-tı¯-za -shun) adaptive immunity A vertebrate-specific Physiological adjustment to a change in an defense that is mediated by B lymphocytes ch chose environmental factor. (B cells) and T lymphocytes (T cells). It e¯ meet acetyl CoA Acetyl coenzyme A; the entry com- exhibits specificity, memory, and self-nonself e/eh bet pound for the citric acid cycle in cellular respi- recognition. Also called acquired immunity. g game ration, formed from a fragment of pyruvate adaptive radiation Period of evolutionary change ı¯ ice attached to a coenzyme. in which groups of organisms form many new i hit acetylcholine (asЈ-uh-til-ko–Ј-le¯n) One of the species whose adaptations allow them to fill dif- ks box most common neurotransmitters; functions by ferent ecological roles in their communities. kw quick binding to receptors and altering the perme- addition rule A rule of probability stating that ng song ability of the postsynaptic membrane to specific the probability of any one of two or more mu- o- robe ions, either depolarizing or hyperpolarizing the tually exclusive events occurring can be deter- membrane. mined by adding their individual probabilities. o ox acid A substance that increases the hydrogen ion adenosine triphosphate See ATP (adenosine oy boy concentration of a solution.