Complement Inhibitor Crig/FH Ameliorates Renal Ischemia Reperfusion Injury Via Activation of PI3K/AKT Signaling
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -
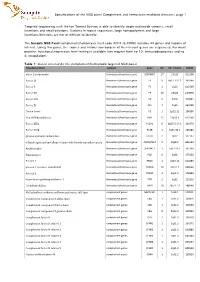
Specifications of the NGS Panel Complement and Hemostasis Mediated Diseases| Page 1
Specifications of the NGS panel Complement and hemostasis mediated diseases| page 1 Targeted sequencing with the Ion Torrent System is able to identify single nucleotide variants, small insertions and small deletions. Variants in repeat sequences, large homopolymers and large insertions/deletions are not or difficult to identify. The Sanquin NGS Panel complement/hemostasis (test code X001 to X006) includes 44 genes and regions of interest. Using this panel, the exones and intron/exon borders of the relevant genes are sequenced. For most proteins, functional/expression level testing is available (see request form no 10: immunodiagnostics and no 4: coagulation). Table 1: Genes covered by the complement/hemostasis targeted NGS panel. Encoded protein Context Gene Chr Chr. Positie OMIM alpha 2-antiplasmin Hemostasis/trombosis gene SERPINF2 17 17p13 613168 Factor IX Hemostasis/trombosis gene F9 X Xq27.1-27.2 300746 Factor V Hemostasis/trombosis gene F5 1 1q23 612309 Factor VII Hemostasis/trombosis gene F7 13 13q34 613878 Factor VIII Hemostasis/trombosis gene F8 X Xq28 300841 Factor XI Hemostasis/trombosis gene F11 4 4q35 264900 Tissue factor Hemostasis/trombosis gene F3 1 1p22-21 134390 Von Willebrand factor Hemostasis/trombosis gene VWF 12 12p13.3 613160 Factor XIIIa Hemostasis/trombosis gene F13A1 6 6p25.3-24.3 134570 Factor XIIIb Hemostasis/trombosis gene F13B 1 1q31-32.1 134580 gamma-glutamyl carboxylase Hemostasis/trombosis gene GGCX 2 2p12 137167 a Disintergrin and metalloproteinase wih thrombospondin repeats Hemostasis/trombosis gene -

Functions of the Complement Components C3 and C5 During Sepsis
The FASEB Journal • Research Communication Functions of the complement components C3 and C5 during sepsis Michael A. Flierl,*,1 Daniel Rittirsch,*,1 Brian A. Nadeau,* Danielle E. Day,* Firas S. Zetoune,* J. Vidya Sarma,* Markus S. Huber-Lang,† and Peter A. Ward*,2 *Department of Pathology, University of Michigan Medical School, Ann Arbor, Michigan, USA; and †Department of Trauma-, Hand- and Reconstructive Surgery, University of Ulm Medical School, Ulm, Germany ABSTRACT Activation of the complement system is a mia (2, 3). Thus, some clinicians preferably refer to this key event in the pathogenesis of sepsis. Nevertheless, complex of symptoms as “sepsis syndrome.” It is of the exact mechanisms remain inadequately understood. concern that doctors have seen a rapid increase in In the current study, we examined the role of comple- hospitalization and mortality rates for severe sepsis in ment C3 and C5 in sepsis in wild-type and C3- or the United States between 1993 and 2003 while mortal- C5-deficient mice induced by cecal ligation and punc- ity rates only slightly decreased (4). During this 11-year ؊/؊ ture. When compared to wild-type mice, C5 showed period, the hospitalization rate has almost doubled and ؊/؊ identical survival, and C3 presented significantly is considerably higher than it has been previously reduced survival. Interestingly, this was associated with predicted, making septicemia now the 10th leading significant decreases in plasma levels of proinflamma- .(؊/؊ cause of death in the United States. (5 tory mediators. Moreover, although septic C3 ani- Encroachment of pathogens prompts the comple- mals displayed a 10-fold increase of blood-borne bac- ؊/؊ ment cascade, which plays a decisive role in the host’s teria, C5 animals exhibited a 400-fold increase in immune response (1, 6). -

Identification of a Novel Mutation in the C6 Gene of a Han Chinese C6SD Child with Meningococcal Disease
EXPERIMENTAL AND THERAPEUTIC MEDICINE 21: 510, 2021 Identification of a novel mutation in the C6 gene of a Han Chinese C6SD child with meningococcal disease AI‑QIAN ZHANG1*, YU‑XING LIU2*, JIE‑YUAN JIN2, CHEN‑YU WANG2, LIANG‑LIANG FAN2,3 and DA‑BAO XU1 1Department of Obstetrics and Gynecology, The Third Xiangya Hospital of Central South University, Changsha, Hunan 410011; 2Department of Cell Biology; 3Hunan Key Laboratory of Animals for Human Disease, School of Life Sciences, Central South University, Changsha, Hunan 410013, P.R. China Received July 18, 2020; Accepted February 5, 2021 DOI: 10.3892/etm.2021.9941 Abstract. Deficiency of the sixth complement component against invading pathogens (2). Genetic deficiencies of any of (C6D) is a genetic disease associated with increased suscep‑ the terminal complement components lead to failure to form tibility to Neisseria meningitides infection. Individuals with the MAC and susceptibility to Neisseria meningitides (Nm) C6D usually present with recurrent meningococcal disease infections, the so‑called terminal complement component (MD). According to the patients' C6 levels, C6D is divided deficiencies (TCCD) (3). into complete genetic deficiency of C6 and subtotal deficiency As one of five terminal complement components, the sixth of C6 (C6SD). The present study reported on a Han Chinese complement component (C6) is a constituent of the MAC. pediatric patient with MD, in whom further investigation Deficiency of C6 (C6D; Online Mendelian Inheritance in Man revealed a C6SD genetic lesion. A heterozygote nonsense #612446) is associated with increased susceptibility to Nm mutation (c.1062C>G/p.Y354*) in the C6 gene was identified infections and patients with C6D usually present with recurrent by Sanger sequencing. -

Complement Component 5 Contributes to Poor Disease Outcome in Humans and Mice with Pneumococcal Meningitis
Complement component 5 contributes to poor disease outcome in humans and mice with pneumococcal meningitis Bianca Woehrl, … , Uwe Koedel, Diederik van de Beek J Clin Invest. 2011;121(10):3943-3953. https://doi.org/10.1172/JCI57522. Research Article Immunology Pneumococcal meningitis is the most common and severe form of bacterial meningitis. Fatality rates are substantial, and long-term sequelae develop in about half of survivors. Disease outcome has been related to the severity of the proinflammatory response in the subarachnoid space. The complement system, which mediates key inflammatory processes, has been implicated as a modulator of pneumococcal meningitis disease severity in animal studies. Additionally, SNPs in genes encoding complement pathway proteins have been linked to susceptibility to pneumococcal infection, although no associations with disease severity or outcome have been established. Here, we have performed a robust prospective nationwide genetic association study in patients with bacterial meningitis and found that a common nonsynonymous complement component 5 (C5) SNP (rs17611) is associated with unfavorable disease outcome. C5 fragment levels in cerebrospinal fluid (CSF) of patients with bacterial meningitis correlated with several clinical indicators of poor prognosis. Consistent with these human data, C5a receptor–deficient mice with pneumococcal meningitis had lower CSF wbc counts and decreased brain damage compared with WT mice. Adjuvant treatment with C5-specific monoclonal antibodies prevented death in all mice with pneumococcal meningitis. Thus, our results suggest C5-specific monoclonal antibodies could be a promising new antiinflammatory adjuvant therapy for pneumococcal meningitis. Find the latest version: https://jci.me/57522/pdf Research article Complement component 5 contributes to poor disease outcome in humans and mice with pneumococcal meningitis Bianca Woehrl,1 Matthijs C. -

Target Deletion of Complement Component 9
www.nature.com/scientificreports OPEN Target deletion of complement component 9 attenuates antibody-mediated hemolysis and Received: 08 March 2016 Accepted: 01 July 2016 lipopolysaccharide (LPS)-induced Published: 22 July 2016 acute shock in mice Xiaoyan Fu1,*, Jiyu Ju1,*, Zhijuan Lin1, Weiling Xiao1, Xiaofang Li1, Baoxiang Zhuang1, Tingting Zhang1, Xiaojun Ma1, Xiangyu Li1, Chao Ma1, Weiliang Su1, Yuqi Wang1, Xuebin Qin2 & Shujuan Liang1 Terminal complement membrane attack complex (MAC) formation is induced initially by C5b, followed by the sequential condensation of the C6, C7, C8. Polymerization of C9 to the C5b-8 complex forms the C5b-9 (or MAC). The C5b-9 forms lytic or non lytic pores in the cell membrane destroys membrane integrity. The biological functionalities of MAC has been previously investigated by using either the mice deficient in C5 and C6, or MAC’s regulator CD59. However, there is no available C9 deficient mice (mC9−/−) for directly dissecting the role of C5b-9 in the pathogenesis of human diseases. Further, since C5b-7 and C5b-8 complexes form non lytic pore, it may also plays biological functionality. To better understand the role of terminal complement cascades, here we report a successful generation of mC9−/−. We demonstrated that lack of C9 attenuates anti-erythrocyte antibody-mediated hemolysis or LPS-induced acute shock. Further, the rescuing effect on the acute shock correlates with the less release of IL-1β in mC9−/−, which is associated with suppression of MAC-mediated inflammasome activation in mC9−/−. Taken together, these results not only confirm the critical role of C5b-9 in complement- mediated hemolysis and but also highlight the critical role of C5b-9 in inflammasome activation. -

A Computational Model for the Evaluation of Complement System Regulation Under Homeostasis, Disease, and Drug Intervention
bioRxiv preprint doi: https://doi.org/10.1101/225029; this version posted November 25, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. A Computational Model for the Evaluation of Complement System Regulation under Homeostasis, Disease, and Drug Intervention Nehemiah Zewde and Dimitrios Morikis* Department of Bioengineering University of California, Riverside *Corresponding author. E-mail address: [email protected] (D. Morikis) Key words: Computational ODE model; immune system regulation; complement system pathways; disease model; Factor H; compstatin; eculizumab. Page 1 of 33 bioRxiv preprint doi: https://doi.org/10.1101/225029; this version posted November 25, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Highlights • Computational model describing dynamics of complement system activation pathways • Complement dysregulation leads to deviation from homeostasis and to inflammatory diseases • Model identifies biomarkers to quantify the effects of complement dysregulation • Known drugs restore impaired dynamics of complement biomarkers under dysregulation • Disease-specific models are suitable for diagnosis and patient-specific drug treatment Page 2 of 33 bioRxiv preprint doi: https://doi.org/10.1101/225029; this version posted November 25, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract The complement system is a part of innate immunity that rapidly removes invading pathogens and impaired host-cells. Activation of the complement system is balanced under homeostasis by regulators that protect healthy host-cells. -

(MDR/TAP), Member 1 ABL1 NM 00
Official Symbol Accession Official Full Name ABCB1 NM_000927.3 ATP-binding cassette, sub-family B (MDR/TAP), member 1 ABL1 NM_005157.3 c-abl oncogene 1, non-receptor tyrosine kinase ADA NM_000022.2 adenosine deaminase AHR NM_001621.3 aryl hydrocarbon receptor AICDA NM_020661.1 activation-induced cytidine deaminase AIRE NM_000383.2 autoimmune regulator APP NM_000484.3 amyloid beta (A4) precursor protein ARG1 NM_000045.2 arginase, liver ARG2 NM_001172.3 arginase, type II ARHGDIB NM_001175.4 Rho GDP dissociation inhibitor (GDI) beta ATG10 NM_001131028.1 ATG10 autophagy related 10 homolog (S. cerevisiae) ATG12 NM_004707.2 ATG12 autophagy related 12 homolog (S. cerevisiae) ATG16L1 NM_198890.2 ATG16 autophagy related 16-like 1 (S. cerevisiae) ATG5 NM_004849.2 ATG5 autophagy related 5 homolog (S. cerevisiae) ATG7 NM_001136031.2 ATG7 autophagy related 7 homolog (S. cerevisiae) ATM NM_000051.3 ataxia telangiectasia mutated B2M NM_004048.2 beta-2-microglobulin B3GAT1 NM_018644.3 beta-1,3-glucuronyltransferase 1 (glucuronosyltransferase P) BATF NM_006399.3 basic leucine zipper transcription factor, ATF-like BATF3 NM_018664.2 basic leucine zipper transcription factor, ATF-like 3 BAX NM_138761.3 BCL2-associated X protein BCAP31 NM_005745.7 B-cell receptor-associated protein 31 BCL10 NM_003921.2 B-cell CLL/lymphoma 10 BCL2 NM_000657.2 B-cell CLL/lymphoma 2 BCL2L11 NM_138621.4 BCL2-like 11 (apoptosis facilitator) BCL3 NM_005178.2 B-cell CLL/lymphoma 3 BCL6 NM_001706.2 B-cell CLL/lymphoma 6 BID NM_001196.2 BH3 interacting domain death agonist BLNK NM_013314.2 -
Complement Inhibition for Improved Nerve Regeneration
(19) TZZ __T (11) EP 2 698 166 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.: of the grant of the patent: A61K 39/395 (2006.01) A61P 25/00 (2006.01) 30.09.2015 Bulletin 2015/40 (21) Application number: 13181901.3 (22) Date of filing: 10.10.2007 (54) Complement inhibition for improved nerve regeneration Komplementhemmung für verbesserte Nervenregeneration Inhibition du complément pour la régénération nerveuse améliorée (84) Designated Contracting States: (56) References cited: AT BE BG CH CY CZ DE DK EE ES FI FR GB GR EP-A1- 1 738 763 WO-A2-00/29033 HU IE IS IT LI LT LU LV MC MT NL PL PT RO SE WO-A2-03/009803 WO-A2-2005/016285 SI SK TR • LI L ET AL: "EFFECTS OF RECOMBINANT SCR1 (30) Priority: 10.10.2006 US 850277 P ON THE IMMUNE INFLAMMATORY REACTION IN ACUTE SPINAL CORD INJURY TISSUE OF (43) Date of publication of application: RATS", ZHONGHUA CHUANGSHANG ZAZHI - 19.02.2014 Bulletin 2014/08 CHINESE JOURNAL OF TRAUMATOLOGY, SICHUAN, CN, vol. 8, no. 1, 1 February 2005 (62) Document number(s) of the earlier application(s) in (2005-02-01), pages 49-53, XP009057804, ISSN: accordance with Art. 76 EPC: 1001-8050 07834628.5 / 2 073 898 • BUREEVA S ET AL: "Drug design using the example of the complement system inhibitors’ (73) Proprietor: Regenesance B.V. development", DRUG DISCOVERY TODAY, 1105 AZ Amsterdam Zuidoost (NL) ELSEVIER, RAHWAY, NJ, US, vol. 10, no. 22, 15 November 2005 (2005-11-15), pages 1535-1542, (72) Inventors: XP027684945, ISSN: 1359-6446 [retrieved on • Baas, Frank 2005-11-15] 1213 VL Hilversum (NL) • WOODRUFF TRENT M ET AL: "Therapeutic • Ramaglia, Valeria activityof C5a receptorantagonists in arat model 1015 MH Amsterdam (NL) of neurodegeneration", FASEB JOURNAL, FED. -

Proteomic Landscape of the Human Choroid–Retinal Pigment Epithelial Complex
Supplementary Online Content Skeie JM, Mahajan VB. Proteomic landscape of the human choroid–retinal pigment epithelial complex. JAMA Ophthalmol. Published online July 24, 2014. doi:10.1001/jamaophthalmol.2014.2065. eFigure 1. Choroid–retinal pigment epithelial (RPE) proteomic analysis pipeline. eFigure 2. Gene ontology (GO) distributions and pathway analysis of human choroid– retinal pigment epithelial (RPE) protein show tissue similarity. eMethods. Tissue collection, mass spectrometry, and analysis. eTable 1. Complete table of proteins identified in the human choroid‐RPE using LC‐ MS/MS. eTable 2. Top 50 signaling pathways in the human choroid‐RPE using MetaCore. eTable 3. Top 50 differentially expressed signaling pathways in the human choroid‐RPE using MetaCore. eTable 4. Differentially expressed proteins in the fovea, macula, and periphery of the human choroid‐RPE. eTable 5. Differentially expressed transcription proteins were identified in foveal, macular, and peripheral choroid‐RPE (p<0.05). eTable 6. Complement proteins identified in the human choroid‐RPE. eTable 7. Proteins associated with age related macular degeneration (AMD). This supplementary material has been provided by the authors to give readers additional information about their work. © 2014 American Medical Association. All rights reserved. 1 Downloaded From: https://jamanetwork.com/ on 09/25/2021 eFigure 1. Choroid–retinal pigment epithelial (RPE) proteomic analysis pipeline. A. The human choroid‐RPE was dissected into fovea, macula, and periphery samples. B. Fractions of proteins were isolated and digested. C. The peptide fragments were analyzed using multi‐dimensional LC‐MS/MS. D. X!Hunter, X!!Tandem, and OMSSA were used for peptide fragment identification. E. Proteins were further analyzed using bioinformatics. -

Genome-Wide Expression Differences in Anti-Vegf and Dexamethasone
www.nature.com/scientificreports OPEN Genome-wide expression differences in anti-Vegf and dexamethasone treatment of Received: 10 November 2016 Accepted: 22 June 2017 inflammatory angiogenesis in the Published: xx xx xxxx rat cornea Pierfrancesco Mirabelli, Anthony Mukwaya , Anton Lennikov, Maria Xeroudaki, Beatrice Peebo, Mira Schaupper & Neil Lagali Angiogenesis as a pathological process in the eye can lead to blindness. In the cornea, suppression of angiogenesis by anti-VEGF treatment is only partially effective while steroids, although effective in treating inflammation and angiogenesis, have broad activity leading to undesirable side effects. In this study, genome-wide expression was investigated in a suture-induced corneal neovascularization model in rats, to investigate factors differentially targeted by dexamethasone and anti-Vegf. Topical treatment with either rat-specific anti-Vegf, dexamethasone, or normal goat IgG (sham) was given to sutured corneas for 48 hours, after which in vivo imaging, tissue processing for RNA microarray, and immunofluorescence were performed. Dexamethasone suppressed limbal vasodilation (P < 0.01) and genes in PI3K-Akt, focal adhesion, and chemokine signaling pathways more effectively than anti-Vegf. The most differentially expressed genes were confirmed by immunofluorescence, qRTPCR and Western blot. Strong suppression of Reg3g and the inflammatory chemokinesCcl2 and Cxcl5 and activation of classical complement pathway factors C1r, C1s, C2, and C3 occurred with dexamethasone treatment, effects absent with anti-Vegf treatment. The genome-wide results obtained in this study provide numerous potential targets for specific blockade of inflammation and angiogenesis in the cornea not addressed by anti-Vegf treatment, as possible alternatives to broad-acting immunosuppressive therapy. Angiogenesis is an essential physiologic process occurring during embryogenesis as well as in adult organisms in the context of wound healing, muscle growth, and the menstrual cycle. -

ESID Online Registry: Diseases and Genes
ESID Online Registry: Diseases and Genes as of 30.07.2015 - Please send ideas and questions to [email protected] Main category Sub category PID Diagnosis Gene Autoinflammatory disorders Blau syndrome Caspase recruitment domain-containing protein 15 deficiency (CARD15) CARD15 CINCA syndrome CINCA syndrome CIAS1 (NLRP3) Familial cold autoinflammatory syndrome Familial cold autoinflammatory syndrome CIAS1 (NLRP3) Familial mediterranean fever Familial mediterranean fever defect MEFV Familial periodic fever Hyper IgD syndrome (MVK) MVK TNF-receptor associated periodic fever syndrome (TRAPS) TNFRSF1A Muckle-Wells syndrome Muckle-Wells syndrome CIAS1 (NLRP3) Other autoinflammatory diseases with known genetic defect Other autoinflammatory diseases with known genetic defect CAD14 HOIL1 IL1RN IL36RN LPIN2 PLCG2 PSMB8 SH3BP2 SLC29A3 Periodic fever, unclassified Periodic fever, unclassified none PFAPA Periodic fever aphtous stomatitis, pharyngitis and adenopathy (PFAPA) none Pyogenic sterile arthritis pyoderma gangrenosum and acne Proline/serine/threonine phosphatase-interacting protein 1 deficiency (PSTPIP1) PSTPIP1 Unclassified autoinflammatory diseases Unclassified autoinflammatory diseases none Combined Atypical Severe Combined Immunodeficiency (Atypical SCID) Atypical Severe Combined Immunodeficiency (Atypical SCID) ADA immunodeficiencies Artemis CARD11 CD3d CD3e CD3z Coronin-1A Gamma C (x-linked) IL21R IL7Ralpha JAK3 PNP RAG1 RAG2 Reticular dysgenesia CD4-deficiency Selective CD4 cell deficiency CD4 CD8-deficiency CD8 deficiency CD8A