ESID Online Registry: Diseases and Genes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

WO 2016/147053 Al 22 September 2016 (22.09.2016) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2016/147053 Al 22 September 2016 (22.09.2016) P O P C T (51) International Patent Classification: (71) Applicant: RESVERLOGIX CORP. [CA/CA]; 300, A61K 31/551 (2006.01) A61P 37/02 (2006.01) 4820 Richard Road Sw, Calgary, AB, T3E 6L1 (CA). A61K 31/517 (2006.01) C07D 239/91 (2006.01) (72) Inventors: WASIAK, Sylwia; 431 Whispering Water (21) International Application Number: Trail, Calgary, AB, T3Z 3V1 (CA). KULIKOWSKI, PCT/IB20 16/000443 Ewelina, B.; 31100 Swift Creek Terrace, Calgary, AB, T3Z 0B7 (CA). HALLIDAY, Christopher, R.A.; 403 (22) International Filing Date: 138-18th Avenue SE, Calgary, AB, T2G 5P9 (CA). GIL- 10 March 2016 (10.03.2016) HAM, Dean; 249 Scenic View Close NW, Calgary, AB, (25) Filing Language: English T3L 1Y5 (CA). (26) Publication Language: English (81) Designated States (unless otherwise indicated, for every kind of national protection available): AE, AG, AL, AM, (30) Priority Data: AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, 62/132,572 13 March 2015 (13.03.2015) US BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, 62/264,768 8 December 2015 (08. 12.2015) US DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, [Continued on nextpage] (54) Title: COMPOSITIONS AND THERAPEUTIC METHODS FOR THE TREATMENT OF COMPLEMENT-ASSOCIATED DISEASES (57) Abstract: The invention comprises methods of modulating the complement cascade in a mammal and for treating and/or preventing diseases and disorders as sociated with the complement pathway by administering a compound of Formula I or Formula II, such as, for example, 2-(4-(2-hydroxyethoxy)-3,5-dimethylphenyl)- 5,7-dimethoxyquinazolin-4(3H)-one or a pharmaceutically acceptable salt thereof. -

Newborn Screening for Severe Combined Immunodeficiency and T-Cell Lymphopenia in California, 2010−2017 George S
Newborn Screening for Severe Combined Immunodeficiency and T-cell Lymphopenia in California, 2010–2017 George S. Amatuni, BS,a,b Robert J. Currier, PhD,a Joseph A. Church, MD,c Tracey Bishop,d Elena Grimbacher,e Alan Anh-Chuong Nguyen, MD,f Rajni Agarwal-Hashmi, MD,g Constantino P. Aznar, PhD,d Manish J. Butte, MD, PhD,h Morton J. Cowan, MD,a Morna J. Dorsey, MD, MMSc,a Christopher C. Dvorak, MD,a Neena Kapoor, MD,c Donald B. Kohn, MD,h M. Louise Markert, MD, PhD,i Theodore B. Moore, MD,h Stanley J. Naides, MD,j Stanley Sciortino, PhD, MPH,d Lisa Feuchtbaum, DrPH, MPH,d Rasoul A. Koupaei, PhD,d Jennifer M. Puck, MDa OBJECTIVES: Newborn screening for severe combined immunodeficiency (SCID) was instituted in abstract California in 2010. In the ensuing 6.5 years, 3 252 156 infants in the state had DNA from dried blood spots assayed for T-cell receptor excision circles (TRECs). Abnormal TREC results were followed-up with liquid blood testing for T-cell abnormalities. We report the performance of the SCID screening program and the outcomes of infants who were identified. METHODS: Data that were reviewed and analyzed included demographics, nursery summaries, TREC and lymphocyte flow-cytometry values, and available follow-up, including clinical and genetic diagnoses, treatments, and outcomes. RESULTS: Infants with clinically significant T-cell lymphopenia (TCL) were successfully identified at a rate of 1 in 15 300 births. Of these, 50 cases of SCID, or 1 in 65 000 births (95% confidence interval 1 in 51 000–1 in 90 000) were found. -

Complement Factor B Antibody Cat
Complement Factor B Antibody Cat. No.: 16-846 Complement Factor B Antibody Immunohistochemistry of paraffin-embedded human mammary cancer using Immunohistochemistry of paraffin-embedded mouse lung using Complement Factor B Complement Factor B antibody (16-846) at dilution of 1:200 (40x lens). antibody (16-846) at dilution of 1:200 (40x lens). Immunofluorescence analysis of Raw264.7 cells using Complement Factor B Polyclonal Antibody (16-846) at dilution of 1:100 (40x lens). Blue: DAPI for nuclear staining. September 29, 2021 1 https://www.prosci-inc.com/complement-factor-b-antibody-16-846.html Specifications HOST SPECIES: Rabbit SPECIES REACTIVITY: Human, Mouse, Rat Recombinant fusion protein containing a sequence corresponding to amino acids 80-420 IMMUNOGEN: of human Complement Factor B (NP_001701.2). TESTED APPLICATIONS: IF, IHC, IP, WB WB: ,1:500 - 1:2000 IHC: ,1:50 - 1:200 APPLICATIONS: IF: ,1:50 - 1:200 IP: ,1:20 - 1:50 POSITIVE CONTROL: 1) HT-29 2) K-562 3) 293T 4) A-549 5) HepG2 6) Mouse liver PREDICTED MOLECULAR Observed: 86kDa WEIGHT: Properties PURIFICATION: Affinity purification CLONALITY: Polyclonal ISOTYPE: IgG CONJUGATE: Unconjugated PHYSICAL STATE: Liquid BUFFER: PBS with 0.02% sodium azide, 50% glycerol, pH7.3. STORAGE CONDITIONS: Store at -20˚C. Avoid freeze / thaw cycles. Additional Info OFFICIAL SYMBOL: CFB September 29, 2021 2 https://www.prosci-inc.com/complement-factor-b-antibody-16-846.html Complement factor B, C3/C5 convertase, Glycine-rich beta glycoprotein, GBG, PBF2, ALTERNATE NAMES: Properdin factor B, Complement factor B Ba fragment, Complement factor B Bb fragment, CFB, BF, BFD GENE ID: 629 USER NOTE: Optimal dilutions for each application to be determined by the researcher. -

Review Article Complement System and Age-Related Macular Degeneration: Implications of Gene-Environment Interaction for Preventive and Personalized Medicine
Hindawi BioMed Research International Volume 2018, Article ID 7532507, 13 pages https://doi.org/10.1155/2018/7532507 Review Article Complement System and Age-Related Macular Degeneration: Implications of Gene-Environment Interaction for Preventive and Personalized Medicine Andrea Maugeri ,1 Martina Barchitta ,1 Maria Grazia Mazzone,2 Francesco Giuliano,2 and Antonella Agodi 1 1 Department of Medical and Surgical Sciences and Advanced Technologies “GF Ingrassia”, University of Catania, Via S. Sofa 87, 95123 Catania, Italy 2SIFI SpA, Research and Development Department, Via Ercole Patti 36, 95025 Catania, Italy Correspondence should be addressed to Antonella Agodi; [email protected] Received 18 May 2018; Accepted 18 July 2018; Published 26 August 2018 Academic Editor: Sajib Chakraborty Copyright © 2018 Andrea Maugeri et al. Tis is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Age-related macular degeneration (AMD) is the most common cause of visual loss in developed countries, with a signifcant economic and social burden on public health. Although genome-wide and gene-candidate studies have been enabled to identify genetic variants in the complement system associated with AMD pathogenesis, the efect of gene-environment interaction is still under debate. In this review we provide an overview of the role of complement system and its genetic variants in AMD, summarizing the consequences of the interaction between genetic and environmental risk factors on AMD onset, progression, and therapeutic response. Finally, we discuss the perspectives of current evidence in the feld of genomics driven personalized medicine and public health. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Practice Parameter for the Diagnosis and Management of Primary Immunodeficiency
Practice parameter Practice parameter for the diagnosis and management of primary immunodeficiency Francisco A. Bonilla, MD, PhD, David A. Khan, MD, Zuhair K. Ballas, MD, Javier Chinen, MD, PhD, Michael M. Frank, MD, Joyce T. Hsu, MD, Michael Keller, MD, Lisa J. Kobrynski, MD, Hirsh D. Komarow, MD, Bruce Mazer, MD, Robert P. Nelson, Jr, MD, Jordan S. Orange, MD, PhD, John M. Routes, MD, William T. Shearer, MD, PhD, Ricardo U. Sorensen, MD, James W. Verbsky, MD, PhD, David I. Bernstein, MD, Joann Blessing-Moore, MD, David Lang, MD, Richard A. Nicklas, MD, John Oppenheimer, MD, Jay M. Portnoy, MD, Christopher R. Randolph, MD, Diane Schuller, MD, Sheldon L. Spector, MD, Stephen Tilles, MD, Dana Wallace, MD Chief Editor: Francisco A. Bonilla, MD, PhD Co-Editor: David A. Khan, MD Members of the Joint Task Force on Practice Parameters: David I. Bernstein, MD, Joann Blessing-Moore, MD, David Khan, MD, David Lang, MD, Richard A. Nicklas, MD, John Oppenheimer, MD, Jay M. Portnoy, MD, Christopher R. Randolph, MD, Diane Schuller, MD, Sheldon L. Spector, MD, Stephen Tilles, MD, Dana Wallace, MD Primary Immunodeficiency Workgroup: Chairman: Francisco A. Bonilla, MD, PhD Members: Zuhair K. Ballas, MD, Javier Chinen, MD, PhD, Michael M. Frank, MD, Joyce T. Hsu, MD, Michael Keller, MD, Lisa J. Kobrynski, MD, Hirsh D. Komarow, MD, Bruce Mazer, MD, Robert P. Nelson, Jr, MD, Jordan S. Orange, MD, PhD, John M. Routes, MD, William T. Shearer, MD, PhD, Ricardo U. Sorensen, MD, James W. Verbsky, MD, PhD GlaxoSmithKline, Merck, and Aerocrine; has received payment for lectures from Genentech/ These parameters were developed by the Joint Task Force on Practice Parameters, representing Novartis, GlaxoSmithKline, and Merck; and has received research support from Genentech/ the American Academy of Allergy, Asthma & Immunology; the American College of Novartis and Merck. -

Differential and Altered Spatial Distribution of Complement Expression in Age-Related Macular Degeneration
Physiology and Pharmacology Differential and Altered Spatial Distribution of Complement Expression in Age-Related Macular Degeneration John T. Demirs, Junzheng Yang, Maura A. Crowley, Michael Twarog, Omar Delgado, Yubin Qiu, Stephen Poor, Dennis S. Rice, Thaddeus P. Dryja,* Karen Anderson,** and Sha-Mei Liao Department of Ophthalmology, Novartis Institutes for Biomedical Research, Cambridge, Massachusetts, United States Correspondence: Sha-Mei Liao, 22 PURPOSE. Dysregulation of the alternative complement pathway is a major pathogenic Windsor Street, Cambridge, MA mechanism in age-related macular degeneration. We investigated whether locally synthe- 02139, USA; sized complement components contribute to AMD by profiling complement expression [email protected]. in postmortem eyes with and without AMD. Current affiliation: *Cogan Eye METHODS. AMD severity grade 1 to 4 was determined by analysis of postmortem acquired Pathology Laboratory, Massachusetts fundus images and hematoxylin and eosin stained histological sections. TaqMan (donor Eye and Ear, Boston, Massachusetts, eyes n = 39) and RNAscope/in situ hybridization (n = 10) were performed to detect United States. = ** Biogen, Cambridge, complement mRNA. Meso scale discovery assay and Western blot (n 31) were used to Massachusetts, United States. measure complement protein levels. Received: September 2, 2020 RESULTS. The levels of complement mRNA and protein expression were approximately Accepted: May 19, 2021 15- to 100-fold (P < 0.0001–0.001) higher in macular retinal pigment epithelium Published: June 23, 2021 (RPE)/choroid tissue than in neural retina, regardless of AMD grade status. Complement Citation: Demirs JT, Yang J, Crowley mRNA and protein levels were modestly elevated in vitreous and the macular neural MA, et al. -

Steffensen Et Al. Supplement 2A
Liver Wild-type Knockout 1 2 4 1 2 4 1 1 1 C C C C C C W W W K K K IMAGE:640919 expressed sequence AA960558 IMAGE:523974 RIKEN cDNA 1810004N01 gene IMAGE:1211217 eukaryotic translation initiation factor 4E binding protein 2 IMAGE:314741 beta-site APP cleaving enzyme IMAGE:1038420 silica-induced gene 41 IMAGE:1229655 soc-2 suppressor of clear homolog C. elegans IMAGE:1150065 Unknown IMAGE:1038486 RIKEN cDNA 2700038M07 gene IMAGE:524351 amyloid beta A4 precursor protein IMAGE:819912 thymus expressed acidic protein IMAGE:779426 RIKEN cDNA 5230400G24 gene IMAGE:945643 ESTs IMAGE:850544 Mpv17 transgene, kidney disease mutant IMAGE:537568 expressed sequence AA675315 IMAGE:1107584 yes-associated protein, 65 kDa IMAGE:961363 expressed sequence AU015422 IMAGE:775218 expressed sequence AI265322 IMAGE:792656 protein phosphatase 1B, magnesium dependent, beta isoform IMAGE:1038592 ESTs, Weakly similar to A43932 mucin 2 precursor, intestinal [H.sapiens] IMAGE:1224917 huntingtin interacting protein 2 IMAGE:751186 ESTs IMAGE:865151 Trk-fused gene IMAGE:523016 SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily a, member 5 IMAGE:765039 FBJ osteosarcoma oncogene B IMAGE:1003995 expressed sequence AI844632 IMAGE:903863 RIKEN cDNA 1300006L01 gene IMAGE:934094 ESTs IMAGE:988962 expressed sequence C77245 IMAGE:1023308 ESTs, Weakly similar to S71512 hypothetical protein T2 - mouse [M.musculus] IMAGE:865317 eukaryotic translation initiation factor 3 IMAGE:720445 ribosomal protein S6 IMAGE:1005417 expressed sequence AU024550 -

Current Perspectives on Primary Immunodeficiency Diseases
Clinical & Developmental Immunology, June–December 2006; 13(2–4): 223–259 Current perspectives on primary immunodeficiency diseases ARVIND KUMAR, SUZANNE S. TEUBER, & M. ERIC GERSHWIN Division of Rheumatology, Allergy and Clinical Immunology, Department of Internal Medicine, University of California at Davis School of Medicine, Davis, CA, USA Abstract Since the original description of X-linked agammaglobulinemia in 1952, the number of independent primary immunodeficiency diseases (PIDs) has expanded to more than 100 entities. By definition, a PID is a genetically determined disorder resulting in enhanced susceptibility to infectious disease. Despite the heritable nature of these diseases, some PIDs are clinically manifested only after prerequisite environmental exposures but they often have associated malignant, allergic, or autoimmune manifestations. PIDs must be distinguished from secondary or acquired immunodeficiencies, which are far more common. In this review, we will place these immunodeficiencies in the context of both clinical and laboratory presentations as well as highlight the known genetic basis. Keywords: Primary immunodeficiency disease, primary immunodeficiency, immunodeficiencies, autoimmune Introduction into a uniform nomenclature (Chapel et al. 2003). The International Union of Immunological Societies Acquired immunodeficiencies may be due to malnu- (IUIS) has subsequently convened an international trition, immunosuppressive or radiation therapies, infections (human immunodeficiency virus, severe committee of experts every two to three years to revise sepsis), malignancies, metabolic disease (diabetes this classification based on new PIDs and further mellitus, uremia, liver disease), loss of leukocytes or understanding of the molecular basis. A recent IUIS immunoglobulins (Igs) via the gastrointestinal tract, committee met in 2003 in Sintra, Portugal with its kidneys, or burned skin, collagen vascular disease such findings published in 2004 in the Journal of Allergy and as systemic lupus erythematosis, splenectomy, and Clinical Immunology (Chapel et al. -
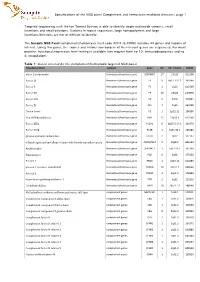
Specifications of the NGS Panel Complement and Hemostasis Mediated Diseases| Page 1
Specifications of the NGS panel Complement and hemostasis mediated diseases| page 1 Targeted sequencing with the Ion Torrent System is able to identify single nucleotide variants, small insertions and small deletions. Variants in repeat sequences, large homopolymers and large insertions/deletions are not or difficult to identify. The Sanquin NGS Panel complement/hemostasis (test code X001 to X006) includes 44 genes and regions of interest. Using this panel, the exones and intron/exon borders of the relevant genes are sequenced. For most proteins, functional/expression level testing is available (see request form no 10: immunodiagnostics and no 4: coagulation). Table 1: Genes covered by the complement/hemostasis targeted NGS panel. Encoded protein Context Gene Chr Chr. Positie OMIM alpha 2-antiplasmin Hemostasis/trombosis gene SERPINF2 17 17p13 613168 Factor IX Hemostasis/trombosis gene F9 X Xq27.1-27.2 300746 Factor V Hemostasis/trombosis gene F5 1 1q23 612309 Factor VII Hemostasis/trombosis gene F7 13 13q34 613878 Factor VIII Hemostasis/trombosis gene F8 X Xq28 300841 Factor XI Hemostasis/trombosis gene F11 4 4q35 264900 Tissue factor Hemostasis/trombosis gene F3 1 1p22-21 134390 Von Willebrand factor Hemostasis/trombosis gene VWF 12 12p13.3 613160 Factor XIIIa Hemostasis/trombosis gene F13A1 6 6p25.3-24.3 134570 Factor XIIIb Hemostasis/trombosis gene F13B 1 1q31-32.1 134580 gamma-glutamyl carboxylase Hemostasis/trombosis gene GGCX 2 2p12 137167 a Disintergrin and metalloproteinase wih thrombospondin repeats Hemostasis/trombosis gene -

ESID Registry – Working Definitions for Clinical Diagnosis of PID
ESID Registry – Working Definitions for Clinical Diagnosis of PID These criteria are only for patients with no genetic diagnosis*. *Exceptions: Atypical SCID, DiGeorge syndrome – a known genetic defect and confirmation of criteria is mandatory. Available entries (Please click on an entry to see the criteria.) Page Acquired angioedema .................................................................................................................................................................. 4 Agammaglobulinemia .................................................................................................................................................................. 4 Asplenia syndrome (Ivemark syndrome) ................................................................................................................................... 4 Ataxia telangiectasia (ATM) ......................................................................................................................................................... 4 Atypical Severe Combined Immunodeficiency (Atypical SCID) ............................................................................................... 5 Autoimmune lymphoproliferative syndrome (ALPS) ................................................................................................................ 5 APECED / APS1 with CMC - Autoimmune polyendocrinopathy candidiasis ectodermal dystrophy (APECED) .................. 5 Barth syndrome ........................................................................................................................................................................... -

Comprehensive Genetic Analysis of Complement and Coagulation Genes in Atypical Hemolytic Uremic Syndrome
BASIC RESEARCH www.jasn.org Comprehensive Genetic Analysis of Complement and Coagulation Genes in Atypical Hemolytic Uremic Syndrome † † † ‡ † Fengxiao Bu,* Tara Maga,* Nicole C. Meyer, Kai Wang, Christie P. Thomas, § † † Carla M. Nester, § and Richard J. H. Smith* § *Interdisciplinary PhD Program in Genetics, †Molecular Otolaryngology and Renal Research Laboratories, ‡Department of Biostatistics, College of Public Health, and §Rare Renal Disease Clinic, Departments of Pediatrics and Internal Medicine, Carver College of Medicine, University of Iowa, Iowa City, Iowa ABSTRACT Atypical hemolytic uremic syndrome (aHUS) is a thrombotic microangiopathy caused by uncontrolled activation of the alternative pathway of complement at the cell surface level that leads to microangiopathic hemolytic anemia, thrombocytopenia, and acute kidney failure. In approximately one half of affected patients, pathogenic loss-of-function variants in regulators of complement or gain-of-function variants in effectors of complement are identified, clearly implicating complement in aHUS. However, there are strong lines of evidence supporting the presence of additional genetic contributions to this disease. To identify novel aHUS-associated genes, we completed a comprehensive screen of the complement and coagulation pathways in 36 patients with sporadic aHUS using targeted genomic enrichment and mas- sively parallel sequencing. After variant calling, quality control, and hard filtering, we identified 84 reported or novel nonsynonymous variants, 22 of which have been previously associated with disease. Using computational prediction methods, 20 of the remaining 62 variants were predicted to be dele- terious. Consistent with published data, nearly one half of these 42 variants (19; 45%) were found in genes implicated in the pathogenesis of aHUS. Several genes in the coagulation pathway were also identified as important in the pathogenesis of aHUS.