Functions of the Complement Components C3 and C5 During Sepsis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

MONONINE (“Difficulty ® Monoclonal Antibody Purified in Concentrating”; Subject Recovered)
CSL Behring IU/kg (n=38), 0.98 ± 0.45 K at doses >95-115 IU/kg (n=21), 0.70 ± 0.38 K at doses >115-135 IU/kg (n=2), 0.67 K at doses >135-155 IU/kg (n=1), and 0.73 ± 0.34 K at doses >155 IU/kg (n=5). Among the 36 subjects who received these high doses, only one (2.8%) Coagulation Factor IX (Human) reported an adverse experience with a possible relationship to MONONINE (“difficulty ® Monoclonal Antibody Purified in concentrating”; subject recovered). In no subjects were thrombo genic complications MONONINE observed or reported.4 only The manufacturing procedure for MONONINE includes multiple processing steps that DESCRIPTION have been designed to reduce the risk of virus transmission. Validation studies of the Coagulation Factor IX (Human), MONONINE® is a sterile, stable, lyophilized concentrate monoclonal antibody (MAb) immunoaffinity chromatography/chemical treatment step and of Factor IX prepared from pooled human plasma and is intended for use in therapy nanofiltration step used in the production of MONONINE doc ument the virus reduction of Factor IX deficiency, known as Hemophilia B or Christmas disease. MONONINE is capacity of the processes employed. These studies were conducted using the rel evant purified of extraneous plasma-derived proteins, including Factors II, VII and X, by use of enveloped and non-enveloped viruses. The results of these virus validation studies utilizing immunoaffinity chromatography. A murine monoclonal antibody to Factor IX is used as an a wide range of viruses with different physicochemical properties are summarized in Table affinity ligand to isolate Factor IX from the source material. -

Coagulation Factors Directly Cleave SARS-Cov-2 Spike and Enhance Viral Entry
bioRxiv preprint doi: https://doi.org/10.1101/2021.03.31.437960; this version posted April 1, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Coagulation factors directly cleave SARS-CoV-2 spike and enhance viral entry. Edward R. Kastenhuber1, Javier A. Jaimes2, Jared L. Johnson1, Marisa Mercadante1, Frauke Muecksch3, Yiska Weisblum3, Yaron Bram4, Robert E. Schwartz4,5, Gary R. Whittaker2 and Lewis C. Cantley1,* Affiliations 1. Meyer Cancer Center, Department of Medicine, Weill Cornell Medical College, New York, NY, USA. 2. Department of Microbiology and Immunology, Cornell University, Ithaca, New York, USA. 3. Laboratory of Retrovirology, The Rockefeller University, New York, NY, USA. 4. Division of Gastroenterology and Hepatology, Department of Medicine, Weill Cornell Medicine, New York, NY, USA. 5. Department of Physiology, Biophysics and Systems Biology, Weill Cornell Medicine, New York, NY, USA. *Correspondence: [email protected] bioRxiv preprint doi: https://doi.org/10.1101/2021.03.31.437960; this version posted April 1, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Summary Coagulopathy is recognized as a significant aspect of morbidity in COVID-19 patients. The clotting cascade is propagated by a series of proteases, including factor Xa and thrombin. Other host proteases, including TMPRSS2, are recognized to be important for cleavage activation of SARS-CoV-2 spike to promote viral entry. Using biochemical and cell-based assays, we demonstrate that factor Xa and thrombin can also directly cleave SARS-CoV-2 spike, enhancing viral entry. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Thrombin-Jmi
THROMBIN-JMI - thrombin, topical (bovine) THROMBIN-JMI; THROMBIN-JMI PUMP SPRAY KIT; THROMBIN-JMI SYRINGE SPRAY KIT - thrombin, topical (bovine) THROMBIN-JMI SYRINGE SPRAY KIT - thrombin, topical (bovine) THROMBIN-JMI EPISTAXIS KIT - thrombin, topical (bovine) King Pharmaceuticals, Inc. ---------- THROMBIN, TOPICAL U.S.P. (BOVINE ORIGIN) THROMBIN-JMI® Thrombin, Topical (Bovine) must not be injected! Apply on the surface of bleeding tissue. DESCRIPTION The thrombin in Thrombin, Topical (Bovine Origin) THROMBIN-JMI® is a protein substance produced through a conversion reaction in which prothrombin of bovine origin is activated by tissue thromboplastin of bovine origin in the presence of calcium chloride. It is supplied as a sterile powder that has been freeze-dried in the final container. Also contained in the preparation are mannitol and sodium chloride. Mannitol is included to make the dried product friable and more readily soluble. The material contains no preservative. THROMBIN-JMI® has been chromatographically purified and further processed by ultrafiltration. Analytical studies demonstrate the current manufacturing process’ capability to remove significant amounts of extraneous proteins, and result in a reduction of Factor Va light chain content to levels below the limit of detection of semi-quantitative Western Blot assay (<92 ng/mL, when reconstituted as directed). The clinical significance of these findings is unknown. CLINICAL PHARMACOLOGY THROMBIN-JMI® requires no intermediate physiological agent for its action. It clots the fibrinogen of the blood directly. Failure to clot blood occurs in the rare case where the primary clotting defect is the absence of fibrinogen itself. The speed with which thrombin clots blood is dependent upon the concentration of both thrombin and fibrinogen. -
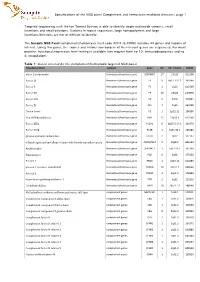
Specifications of the NGS Panel Complement and Hemostasis Mediated Diseases| Page 1
Specifications of the NGS panel Complement and hemostasis mediated diseases| page 1 Targeted sequencing with the Ion Torrent System is able to identify single nucleotide variants, small insertions and small deletions. Variants in repeat sequences, large homopolymers and large insertions/deletions are not or difficult to identify. The Sanquin NGS Panel complement/hemostasis (test code X001 to X006) includes 44 genes and regions of interest. Using this panel, the exones and intron/exon borders of the relevant genes are sequenced. For most proteins, functional/expression level testing is available (see request form no 10: immunodiagnostics and no 4: coagulation). Table 1: Genes covered by the complement/hemostasis targeted NGS panel. Encoded protein Context Gene Chr Chr. Positie OMIM alpha 2-antiplasmin Hemostasis/trombosis gene SERPINF2 17 17p13 613168 Factor IX Hemostasis/trombosis gene F9 X Xq27.1-27.2 300746 Factor V Hemostasis/trombosis gene F5 1 1q23 612309 Factor VII Hemostasis/trombosis gene F7 13 13q34 613878 Factor VIII Hemostasis/trombosis gene F8 X Xq28 300841 Factor XI Hemostasis/trombosis gene F11 4 4q35 264900 Tissue factor Hemostasis/trombosis gene F3 1 1p22-21 134390 Von Willebrand factor Hemostasis/trombosis gene VWF 12 12p13.3 613160 Factor XIIIa Hemostasis/trombosis gene F13A1 6 6p25.3-24.3 134570 Factor XIIIb Hemostasis/trombosis gene F13B 1 1q31-32.1 134580 gamma-glutamyl carboxylase Hemostasis/trombosis gene GGCX 2 2p12 137167 a Disintergrin and metalloproteinase wih thrombospondin repeats Hemostasis/trombosis gene -

Protein C Product Monograph 1995 COAMATIC® Protein C Protein C
Protein C Product Monograph 1995 COAMATIC® Protein C Protein C Protein C, Product Monograph 1995 Frank Axelsson, Product Information Manager Copyright © 1995 Chromogenix AB. Version 1.1 Taljegårdsgatan 3, S-431 53 Mölndal, Sweden. Tel: +46 31 706 20 00, Fax: +46 31 86 46 26, E-mail: [email protected], Internet: www.chromogenix.se COAMATIC® Protein C Protein C Contents Page Preface 2 Introduction 4 Determination of protein C activity with 4 snake venom and S-2366 Biochemistry 6 Protein C biochemistry 6 Clinical Aspects 10 Protein C deficiency 10 Assay Methods 13 Protein C assays 13 Laboratory aspects 16 Products 17 Diagnostic kits from Chromogenix 17 General assay procedure 18 COAMATIC® Protein C 19 References 20 Glossary 23 3 Protein C, version 1.1 Preface The blood coagulation system is carefully controlled in vivo by several anticoagulant mechanisms, which ensure that clot propagation does not lead to occlusion of the vasculature. The protein C pathway is one of these anticoagulant systems. During the last few years it has been found that inherited defects of the protein C system are underlying risk factors in a majority of cases with familial thrombophilia. The factor V gene mutation recently identified in conjunction with APC resistance is such a defect which, in combination with protein C deficiency, increases the thrombosis risk considerably. The Chromogenix Monographs [Protein C and APC-resistance] give a didactic and illustrated picture of the protein C environment by presenting a general view of medical as well as technical matters. They serve as an excellent introduction and survey to everyone who wishes to learn quickly about this field of medicine. -

EXPERT COMMITTEE on BIOLOGICAL STANDARDIZATION Geneva, 17 to 21 October 2016
WHO/BS/2016.2282 ENGLISH ONLY EXPERT COMMITTEE ON BIOLOGICAL STANDARDIZATION Geneva, 17 to 21 October 2016 An international collaborative study to calibrate the WHO 2nd International Standard for Ancrod (15/106) and the WHO Reference Reagent for Batroxobin (15/140) Craig Thelwell1ᶲ, Colin Longstaff1ᶿ, Peter Rigsby2, Matthew Locke1 and Sally Bevan1 1Biotherapeutics Group, Haemostasis Section and 2Biostatistics Section, National Institute for Biological Standards and Control, South Mimms, Herts EN6 3QG, UK ᶲProject leader for Ancrod; ᶿProject leader for Batroxobin NOTE: This document has been prepared for the purpose of inviting comments and suggestions on the proposals contained therein, which will then be considered by the Expert Committee on Biological Standardization (ECBS). Comments MUST be received by 16 September 2016 and should be addressed to the World Health Organization, 1211 Geneva 27, Switzerland, attention: Technologies, Standards and Norms (TSN). Comments may also be submitted electronically to the Responsible Officer: Dr C M Nübling at email: [email protected] © World Health Organization 2016 All rights reserved. Publications of the World Health Organization are available on the WHO web site (www.who.int) or can be purchased from WHO Press, World Health Organization, 20 Avenue Appia, 1211 Geneva 27, Switzerland (tel.: +41 22 791 3264; fax: +41 22 791 4857; e-mail: [email protected]). Requests for permission to reproduce or translate WHO publications – whether for sale or for noncommercial distribution – should be addressed to WHO Press through the WHO web site: (http://www.who.int/about/licensing/copyright_form/en/index.html). The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the World Health Organization concerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. -

Replacing the First Epidermal Growth Factor-Like Domain of Factor IX with That of Factor VII Enhances Activity in Vitro and in Canine Hemophilia B
Replacing the first epidermal growth factor-like domain of factor IX with that of factor VII enhances activity in vitro and in canine hemophilia B. J Y Chang, … , K M Brinkhous, H R Roberts J Clin Invest. 1997;100(4):886-892. https://doi.org/10.1172/JCI119604. Research Article Using the techniques of molecular biology, we made a chimeric Factor IX by replacing the first epidermal growth factor- like domain with that of Factor VII. The resulting recombinant chimeric molecule, Factor IXVIIEGF1, had at least a twofold increase in functional activity in the one-stage clotting assay when compared to recombinant wild-type Factor IX. The increased activity was not due to contamination with activated Factor IX, nor was it due to an increased rate of activation by Factor VIIa-tissue factor or by Factor XIa. Rather, the increased activity was due to a higher affinity of Factor IXVIIEGF1 for Factor VIIIa with a Kd for Factor VIIIa about one order of magnitude lower than that of recombinant wild- type Factor IXa. In addition, results from animal studies show that this chimeric Factor IX, when infused into a dog with hemophilia B, exhibits a greater than threefold increase in clotting activity, and has a biological half-life equivalent to recombinant wild-type Factor IX. Find the latest version: https://jci.me/119604/pdf Replacing the First Epidermal Growth Factor-like Domain of Factor IX with That of Factor VII Enhances Activity In Vitro and in Canine Hemophilia B Jen-Yea Chang,* Dougald M. Monroe,* Darrel W. Stafford,*‡ Kenneth M. -

Assessing Plasmin Generation in Health and Disease
International Journal of Molecular Sciences Review Assessing Plasmin Generation in Health and Disease Adam Miszta 1,* , Dana Huskens 1, Demy Donkervoort 1, Molly J. M. Roberts 1, Alisa S. Wolberg 2 and Bas de Laat 1 1 Synapse Research Institute, 6217 KD Maastricht, The Netherlands; [email protected] (D.H.); [email protected] (D.D.); [email protected] (M.J.M.R.); [email protected] (B.d.L.) 2 Department of Pathology and Laboratory Medicine and UNC Blood Research Center, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA; [email protected] * Correspondence: [email protected]; Tel.: +31-(0)-433030693 Abstract: Fibrinolysis is an important process in hemostasis responsible for dissolving the clot during wound healing. Plasmin is a central enzyme in this process via its capacity to cleave fibrin. The ki- netics of plasmin generation (PG) and inhibition during fibrinolysis have been poorly understood until the recent development of assays to quantify these metrics. The assessment of plasmin kinetics allows for the identification of fibrinolytic dysfunction and better understanding of the relationships between abnormal fibrin dissolution and disease pathogenesis. Additionally, direct measurement of the inhibition of PG by antifibrinolytic medications, such as tranexamic acid, can be a useful tool to assess the risks and effectiveness of antifibrinolytic therapy in hemorrhagic diseases. This review provides an overview of available PG assays to directly measure the kinetics of plasmin formation and inhibition in human and mouse plasmas and focuses on their applications in defining the role of plasmin in diseases, including angioedema, hemophilia, rare bleeding disorders, COVID- 19, or diet-induced obesity. -

Identification of a Novel Mutation in the C6 Gene of a Han Chinese C6SD Child with Meningococcal Disease
EXPERIMENTAL AND THERAPEUTIC MEDICINE 21: 510, 2021 Identification of a novel mutation in the C6 gene of a Han Chinese C6SD child with meningococcal disease AI‑QIAN ZHANG1*, YU‑XING LIU2*, JIE‑YUAN JIN2, CHEN‑YU WANG2, LIANG‑LIANG FAN2,3 and DA‑BAO XU1 1Department of Obstetrics and Gynecology, The Third Xiangya Hospital of Central South University, Changsha, Hunan 410011; 2Department of Cell Biology; 3Hunan Key Laboratory of Animals for Human Disease, School of Life Sciences, Central South University, Changsha, Hunan 410013, P.R. China Received July 18, 2020; Accepted February 5, 2021 DOI: 10.3892/etm.2021.9941 Abstract. Deficiency of the sixth complement component against invading pathogens (2). Genetic deficiencies of any of (C6D) is a genetic disease associated with increased suscep‑ the terminal complement components lead to failure to form tibility to Neisseria meningitides infection. Individuals with the MAC and susceptibility to Neisseria meningitides (Nm) C6D usually present with recurrent meningococcal disease infections, the so‑called terminal complement component (MD). According to the patients' C6 levels, C6D is divided deficiencies (TCCD) (3). into complete genetic deficiency of C6 and subtotal deficiency As one of five terminal complement components, the sixth of C6 (C6SD). The present study reported on a Han Chinese complement component (C6) is a constituent of the MAC. pediatric patient with MD, in whom further investigation Deficiency of C6 (C6D; Online Mendelian Inheritance in Man revealed a C6SD genetic lesion. A heterozygote nonsense #612446) is associated with increased susceptibility to Nm mutation (c.1062C>G/p.Y354*) in the C6 gene was identified infections and patients with C6D usually present with recurrent by Sanger sequencing. -
Thrombin, Topical (Human) • Potential Risk of Thrombosis If Absorbed Systemically (5.2, 11)
HIGHLIGHTS OF PRESCRIBING INFORMATION WARNINGS AND PRECAUTIONS • May carry a risk of transmitting infectious agents such as viruses and These highlights do not include all the information needed to use theoretically, the Creutzfeldt-Jakob disease (CJD) agent, despite EVITHROM safely and effectively. See full prescribing information. manufacturing steps designed to reduce the risk of viral transmission (11). EVITHROM* Thrombin, Topical (Human) • Potential risk of thrombosis if absorbed systemically (5.2, 11). For Topical Use Only ADVERSE REACTIONS Initial U.S approval: 2007 • Anaphylactic reactions may occur (6). INDICATIONS AND USAGE • Adverse events were reported in the clinical trial with similar frequency • As an aid to hemostasis whenever oozing blood and minor bleeding in the two study groups (EVITHROM or bovine thrombin group). The from capillaries and small venules is accessible and control of bleeding most common adverse event reported was procedural complications by standard surgical techniques is ineffective or impractical (1). and pruritus (6). None of the adverse events reported was considered • May be used in conjunction with an Absorbable Gelatin Sponge, USP (1). causally related to EVITHROM administration. • Immunogenicity was evaluated by testing for the development of DOSAGE AND ADMINISTRATION antibodies to highly purified antigens: human thrombin, human Factor • For topical use only. DO NOT INJECT (2.2). V/Va, bovine thrombin and bovine Factor V/Va. None of the patients • The amount required depends upon the area of tissue to be treated treated with EVITHROM developed antibodies to human thrombin or and the method of application. In clinical studies, volumes up to 10 ml to human Factor V/Va. -

Reelin Signals Through Apolipoprotein E Receptor 2 and Cdc42 to Increase Growth Cone Motility and Filopodia Formation
The Journal of Neuroscience, November 3, 2010 • 30(44):14759–14772 • 14759 Cellular/Molecular Reelin Signals through Apolipoprotein E Receptor 2 and Cdc42 to Increase Growth Cone Motility and Filopodia Formation Jost Leemhuis,1,2 Elisabeth Bouche´,1,3 Michael Frotscher,1,4 Frank Henle,1,2 Lutz Hein,2 Joachim Herz,1,6 Dieter K. Meyer,1,2 Marina Pichler,1,2 Gu¨nter Roth,5 Carsten Schwan,1,2 and Hans H. Bock1,3 1Center for Neuroscience, 2Institute of Experimental and Clinical Pharmacology and Toxicology, 3Department of Medicine II, 4Institute of Anatomy and Cell Biology, and 5Department of Microsystems Engineering, Albert-Ludwigs-University, 79104 Freiburg, Germany, and 6Department of Molecular Genetics, University of Texas Southwestern Medical Center, Dallas, Texas 75390-9046 Lipoprotein receptor signaling regulates the positioning and differentiation of postmitotic neurons during development and modulates neuronal plasticity in the mature brain. Depending on the contextual situation, the lipoprotein receptor ligand Reelin can have opposing effects on cortical neurons. We show that Reelin increases growth cone motility and filopodia formation, and identify the underlying signaling cascade. Reelin activates the Rho GTPase Cdc42, known for its role in neuronal morphogenesis and directed migration, in an apolipoprotein E receptor 2-, Disabled-1-, and phosphatidylinositol 3-kinase-dependent manner. We demonstrate that neuronal vesicle trafficking, a Cdc42-controlled process, is increased after Reelin treatment and further provide evidence that the peptidergic VIP/ PACAP38 system and Reelin can functionally interact to promote axonal branching. In conclusion, Reelin-induced activation of Cdc42 contributes to the regulation of the cytoskeleton of individual responsive neurons and converges with other signaling cascades to orchestrate Rho GTPase activity and promote neuronal development.