Target Deletion of Complement Component 9
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -
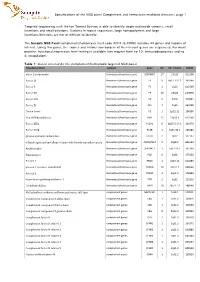
Specifications of the NGS Panel Complement and Hemostasis Mediated Diseases| Page 1
Specifications of the NGS panel Complement and hemostasis mediated diseases| page 1 Targeted sequencing with the Ion Torrent System is able to identify single nucleotide variants, small insertions and small deletions. Variants in repeat sequences, large homopolymers and large insertions/deletions are not or difficult to identify. The Sanquin NGS Panel complement/hemostasis (test code X001 to X006) includes 44 genes and regions of interest. Using this panel, the exones and intron/exon borders of the relevant genes are sequenced. For most proteins, functional/expression level testing is available (see request form no 10: immunodiagnostics and no 4: coagulation). Table 1: Genes covered by the complement/hemostasis targeted NGS panel. Encoded protein Context Gene Chr Chr. Positie OMIM alpha 2-antiplasmin Hemostasis/trombosis gene SERPINF2 17 17p13 613168 Factor IX Hemostasis/trombosis gene F9 X Xq27.1-27.2 300746 Factor V Hemostasis/trombosis gene F5 1 1q23 612309 Factor VII Hemostasis/trombosis gene F7 13 13q34 613878 Factor VIII Hemostasis/trombosis gene F8 X Xq28 300841 Factor XI Hemostasis/trombosis gene F11 4 4q35 264900 Tissue factor Hemostasis/trombosis gene F3 1 1p22-21 134390 Von Willebrand factor Hemostasis/trombosis gene VWF 12 12p13.3 613160 Factor XIIIa Hemostasis/trombosis gene F13A1 6 6p25.3-24.3 134570 Factor XIIIb Hemostasis/trombosis gene F13B 1 1q31-32.1 134580 gamma-glutamyl carboxylase Hemostasis/trombosis gene GGCX 2 2p12 137167 a Disintergrin and metalloproteinase wih thrombospondin repeats Hemostasis/trombosis gene -

ESID Registry – Working Definitions for Clinical Diagnosis of PID
ESID Registry – Working Definitions for Clinical Diagnosis of PID These criteria are only for patients with no genetic diagnosis*. *Exceptions: Atypical SCID, DiGeorge syndrome – a known genetic defect and confirmation of criteria is mandatory. Available entries (Please click on an entry to see the criteria.) Page Acquired angioedema .................................................................................................................................................................. 4 Agammaglobulinemia .................................................................................................................................................................. 4 Asplenia syndrome (Ivemark syndrome) ................................................................................................................................... 4 Ataxia telangiectasia (ATM) ......................................................................................................................................................... 4 Atypical Severe Combined Immunodeficiency (Atypical SCID) ............................................................................................... 5 Autoimmune lymphoproliferative syndrome (ALPS) ................................................................................................................ 5 APECED / APS1 with CMC - Autoimmune polyendocrinopathy candidiasis ectodermal dystrophy (APECED) .................. 5 Barth syndrome ........................................................................................................................................................................... -

Comprehensive Genetic Analysis of Complement and Coagulation Genes in Atypical Hemolytic Uremic Syndrome
BASIC RESEARCH www.jasn.org Comprehensive Genetic Analysis of Complement and Coagulation Genes in Atypical Hemolytic Uremic Syndrome † † † ‡ † Fengxiao Bu,* Tara Maga,* Nicole C. Meyer, Kai Wang, Christie P. Thomas, § † † Carla M. Nester, § and Richard J. H. Smith* § *Interdisciplinary PhD Program in Genetics, †Molecular Otolaryngology and Renal Research Laboratories, ‡Department of Biostatistics, College of Public Health, and §Rare Renal Disease Clinic, Departments of Pediatrics and Internal Medicine, Carver College of Medicine, University of Iowa, Iowa City, Iowa ABSTRACT Atypical hemolytic uremic syndrome (aHUS) is a thrombotic microangiopathy caused by uncontrolled activation of the alternative pathway of complement at the cell surface level that leads to microangiopathic hemolytic anemia, thrombocytopenia, and acute kidney failure. In approximately one half of affected patients, pathogenic loss-of-function variants in regulators of complement or gain-of-function variants in effectors of complement are identified, clearly implicating complement in aHUS. However, there are strong lines of evidence supporting the presence of additional genetic contributions to this disease. To identify novel aHUS-associated genes, we completed a comprehensive screen of the complement and coagulation pathways in 36 patients with sporadic aHUS using targeted genomic enrichment and mas- sively parallel sequencing. After variant calling, quality control, and hard filtering, we identified 84 reported or novel nonsynonymous variants, 22 of which have been previously associated with disease. Using computational prediction methods, 20 of the remaining 62 variants were predicted to be dele- terious. Consistent with published data, nearly one half of these 42 variants (19; 45%) were found in genes implicated in the pathogenesis of aHUS. Several genes in the coagulation pathway were also identified as important in the pathogenesis of aHUS. -

Hypoxia Restrains the Expression of Complement Component 9 in Tumor
Li et al. Cell Death Discovery (2018) 4:63 DOI 10.1038/s41420-018-0064-3 Cell Death Discovery ARTICLE Open Access Hypoxia restrains the expression of complement component 9 in tumor- associated macrophages promoting non- smallcelllungcancerprogression Lei Li1, Hong Yang1,2,3, Yan Li1, Xiao-Dong Li1,3, Ting-Ting Zeng1,Su-XiaLin4,Ying-HuiZhu1 and Xin-Yuan Guan 1,5 Abstract The tumor microenvironment, including stroma cells, signaling molecules, and the extracellular matrix, critically regulates the growth and survival of cancer cells. Dissecting the active molecules in tumor microenvironment may uncover the key factors that can impact cancer progression. Human NSCLC tumor tissue-conditioned medium (TCM) and adjacent nontumor tissue-conditioned medium (NCM) were used to treat two NSCLC cells LSC1 and LAC1, respectively. Cell growth and foci formation assays were applied to assess the effects of TCM and NCM on cancer cells. The active factors were identified by protein mass spectrometry. Cell growth and foci formation assays showed that 8 of 26 NCM and none of TCM could effectively lead to tumor cell lysis, which was known as tumoricidal activity. And then protein mass spectrometry analysis and functional verifications confirmed that complement component 9 (C9) played a crucial role in the complement-dependent cytotoxicity (CDC)-mediated tumoricidal activity in vitro. Furthermore, immunofluorescent staining revealed that C9 specifically expressed in most alveolar macrophages (AMs) 1234567890():,; 1234567890():,; 1234567890():,; 1234567890():,; in adjacent lung tissues and a small fraction of tumor-associated macrophages (TAMs) in NSCLC tissues. Most importantly, the percentage of C9-positive cells in AMs or TAMs was responsible for the tumoricidal activity of NCM and TCM. -

Functions of the Complement Components C3 and C5 During Sepsis
The FASEB Journal • Research Communication Functions of the complement components C3 and C5 during sepsis Michael A. Flierl,*,1 Daniel Rittirsch,*,1 Brian A. Nadeau,* Danielle E. Day,* Firas S. Zetoune,* J. Vidya Sarma,* Markus S. Huber-Lang,† and Peter A. Ward*,2 *Department of Pathology, University of Michigan Medical School, Ann Arbor, Michigan, USA; and †Department of Trauma-, Hand- and Reconstructive Surgery, University of Ulm Medical School, Ulm, Germany ABSTRACT Activation of the complement system is a mia (2, 3). Thus, some clinicians preferably refer to this key event in the pathogenesis of sepsis. Nevertheless, complex of symptoms as “sepsis syndrome.” It is of the exact mechanisms remain inadequately understood. concern that doctors have seen a rapid increase in In the current study, we examined the role of comple- hospitalization and mortality rates for severe sepsis in ment C3 and C5 in sepsis in wild-type and C3- or the United States between 1993 and 2003 while mortal- C5-deficient mice induced by cecal ligation and punc- ity rates only slightly decreased (4). During this 11-year ؊/؊ ture. When compared to wild-type mice, C5 showed period, the hospitalization rate has almost doubled and ؊/؊ identical survival, and C3 presented significantly is considerably higher than it has been previously reduced survival. Interestingly, this was associated with predicted, making septicemia now the 10th leading significant decreases in plasma levels of proinflamma- .(؊/؊ cause of death in the United States. (5 tory mediators. Moreover, although septic C3 ani- Encroachment of pathogens prompts the comple- mals displayed a 10-fold increase of blood-borne bac- ؊/؊ ment cascade, which plays a decisive role in the host’s teria, C5 animals exhibited a 400-fold increase in immune response (1, 6). -

Identification of a Novel Mutation in the C6 Gene of a Han Chinese C6SD Child with Meningococcal Disease
EXPERIMENTAL AND THERAPEUTIC MEDICINE 21: 510, 2021 Identification of a novel mutation in the C6 gene of a Han Chinese C6SD child with meningococcal disease AI‑QIAN ZHANG1*, YU‑XING LIU2*, JIE‑YUAN JIN2, CHEN‑YU WANG2, LIANG‑LIANG FAN2,3 and DA‑BAO XU1 1Department of Obstetrics and Gynecology, The Third Xiangya Hospital of Central South University, Changsha, Hunan 410011; 2Department of Cell Biology; 3Hunan Key Laboratory of Animals for Human Disease, School of Life Sciences, Central South University, Changsha, Hunan 410013, P.R. China Received July 18, 2020; Accepted February 5, 2021 DOI: 10.3892/etm.2021.9941 Abstract. Deficiency of the sixth complement component against invading pathogens (2). Genetic deficiencies of any of (C6D) is a genetic disease associated with increased suscep‑ the terminal complement components lead to failure to form tibility to Neisseria meningitides infection. Individuals with the MAC and susceptibility to Neisseria meningitides (Nm) C6D usually present with recurrent meningococcal disease infections, the so‑called terminal complement component (MD). According to the patients' C6 levels, C6D is divided deficiencies (TCCD) (3). into complete genetic deficiency of C6 and subtotal deficiency As one of five terminal complement components, the sixth of C6 (C6SD). The present study reported on a Han Chinese complement component (C6) is a constituent of the MAC. pediatric patient with MD, in whom further investigation Deficiency of C6 (C6D; Online Mendelian Inheritance in Man revealed a C6SD genetic lesion. A heterozygote nonsense #612446) is associated with increased susceptibility to Nm mutation (c.1062C>G/p.Y354*) in the C6 gene was identified infections and patients with C6D usually present with recurrent by Sanger sequencing. -

The Effect of Bee Venom Peptides Melittin, Tertiapin, and Apamin On
H OH metabolites OH Article The Effect of Bee Venom Peptides Melittin, Tertiapin, and Apamin on the Human Erythrocytes Ghosts: A Preliminary Study 1, 2, 1 3 Agata Swiatły-Błaszkiewicz´ y, Lucyna Mrówczy ´nska y, Eliza Matuszewska , Jan Lubawy , Arkadiusz Urba ´nski 3 , Zenon J. Kokot 1, Grzegorz Rosi ´nski 3 and Jan Matysiak 1,* 1 Department of Inorganic and Analytical Chemistry, Poznan University of Medical Sciences, 60-780 Poznan, Poland; [email protected] (A.S.-B.);´ [email protected] (E.M.); [email protected] (Z.J.K.) 2 Department of Cell Biology, Faculty of Biology, Adam Mickiewicz University in Poznan, 61-614 Poznan, Poland; [email protected] 3 Department of Animal Physiology and Development, Faculty of Biology, Adam Mickiewicz University in Poznan, 61-614 Poznan, Poland; [email protected] (J.L.); [email protected] (A.U.); [email protected] (G.R.) * Correspondence: [email protected] These two authors contributed equally to this work. y Received: 11 April 2020; Accepted: 11 May 2020; Published: 13 May 2020 Abstract: Red blood cells (RBCs) are the most abundant cells in the human blood that have been extensively studied under morphology, ultrastructure, biochemical and molecular functions. Therefore, RBCs are excellent cell models in the study of biologically active compounds like drugs and toxins on the structure and function of the cell membrane. The aim of the present study was to explore erythrocyte ghost’s proteome to identify changes occurring under the influence of three bee venom peptides-melittin, tertiapin, and apamin. We conducted preliminary experiments on the erythrocyte ghosts incubated with these peptides at their non-hemolytic concentrations. -

Complement Component 5 Contributes to Poor Disease Outcome in Humans and Mice with Pneumococcal Meningitis
Complement component 5 contributes to poor disease outcome in humans and mice with pneumococcal meningitis Bianca Woehrl, … , Uwe Koedel, Diederik van de Beek J Clin Invest. 2011;121(10):3943-3953. https://doi.org/10.1172/JCI57522. Research Article Immunology Pneumococcal meningitis is the most common and severe form of bacterial meningitis. Fatality rates are substantial, and long-term sequelae develop in about half of survivors. Disease outcome has been related to the severity of the proinflammatory response in the subarachnoid space. The complement system, which mediates key inflammatory processes, has been implicated as a modulator of pneumococcal meningitis disease severity in animal studies. Additionally, SNPs in genes encoding complement pathway proteins have been linked to susceptibility to pneumococcal infection, although no associations with disease severity or outcome have been established. Here, we have performed a robust prospective nationwide genetic association study in patients with bacterial meningitis and found that a common nonsynonymous complement component 5 (C5) SNP (rs17611) is associated with unfavorable disease outcome. C5 fragment levels in cerebrospinal fluid (CSF) of patients with bacterial meningitis correlated with several clinical indicators of poor prognosis. Consistent with these human data, C5a receptor–deficient mice with pneumococcal meningitis had lower CSF wbc counts and decreased brain damage compared with WT mice. Adjuvant treatment with C5-specific monoclonal antibodies prevented death in all mice with pneumococcal meningitis. Thus, our results suggest C5-specific monoclonal antibodies could be a promising new antiinflammatory adjuvant therapy for pneumococcal meningitis. Find the latest version: https://jci.me/57522/pdf Research article Complement component 5 contributes to poor disease outcome in humans and mice with pneumococcal meningitis Bianca Woehrl,1 Matthijs C. -

Aberrant Complement System Activation in Neurological Disorders
International Journal of Molecular Sciences Review Aberrant Complement System Activation in Neurological Disorders Karolina Ziabska , Malgorzata Ziemka-Nalecz, Paulina Pawelec, Joanna Sypecka and Teresa Zalewska * Mossakowski Medical Research Centre, NeuroRepair Department, Polish Academy of Sciences, 5 Pawinskiego Street, 02-106 Warsaw, Poland; [email protected] (K.Z.); [email protected] (M.Z.-N.); [email protected] (P.P.); [email protected] (J.S.) * Correspondence: [email protected]; Tel.: +48-(22)-608-65-29; Fax: +48-(22)-608-66-23 Abstract: The complement system is an assembly of proteins that collectively participate in the functions of the healthy and diseased brain. The complement system plays an important role in the maintenance of uninjured (healthy) brain homeostasis, contributing to the clearance of invading pathogens and apoptotic cells, and limiting the inflammatory immune response. However, overacti- vation or underregulation of the entire complement cascade within the brain may lead to neuronal damage and disturbances in brain function. During the last decade, there has been a growing interest in the role that this cascading pathway plays in the neuropathology of a diverse array of brain disorders (e.g., acute neurotraumatic insult, chronic neurodegenerative diseases, and psychiatric disturbances) in which interruption of neuronal homeostasis triggers complement activation. Dys- function of the complement promotes a disease-specific response that may have either beneficial or detrimental effects. Despite recent advances, the explicit link between complement component regulation and brain disorders remains unclear. Therefore, a comprehensible understanding of such relationships at different stages of diseases could provide new insight into potential therapeutic targets to ameliorate or slow progression of currently intractable disorders in the nervous system. -

Deficiency of Complement Defense Protein CD59 May Contribute To
The Journal of Neuroscience, October 15, 2000, 20(20):7505–7509 Deficiency of Complement Defense Protein CD59 May Contribute to Neurodegeneration in Alzheimer’s Disease Li-Bang Yang,1 Rena Li,1 Seppo Meri,2 Joseph Rogers,1 and Yong Shen1 1L. J. Roberts Center for Alzheimer’s Research, Sun Health Research Institute, Sun City, Arizona 85351, and 2Department of Immunology, Haartman Institute, University of Helsinki, Finland Complement defense 59 (CD59) is a cell surface glycophos- To evaluate the pathophysiological significance of CD59 alter- phoinositol (GPI)-anchored protein that prevents complement ations in neurons, we exposed cultured NT2 cells, which normally membrane attack complex (MAC) assembly. Here, we present underexpress CD59, and NT2 cells transfected to overexpress evidence from ELISA assays that CD59 protein levels are signif- CD59 to homologous human serum. Lactic acid dehydrogenase icantly decreased in the frontal cortex and hippocampus of assays revealed significant complement-induced cell lysis in CD59- Alzheimer’s disease (AD) compared with nondemented elderly underexpressing NT2 cells and significant protection from such (ND) patients, whereas complement component 9, a final com- lysis in CD59-overexpressing NT2 cells. Moreover, cells expressing ponent to form MAC, is significantly increased. To further confirm normal levels of CD59 showed no evidence of MAC assembly or the CD59 deficit, PI-specific phospholipase C (PIPLC) was used damage after exposure to homologous serum, whereas pretreat- to cleave the CD59 GPI anchor at the cell surface in intact slices ment of these cells with a CD59-neutralizing antibody resulted in from AD and ND cortex. CD59 released by PIPLC cleavage was MAC assembly at the cell surface and morphological damage. -

A Computational Model for the Evaluation of Complement System Regulation Under Homeostasis, Disease, and Drug Intervention
bioRxiv preprint doi: https://doi.org/10.1101/225029; this version posted November 25, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. A Computational Model for the Evaluation of Complement System Regulation under Homeostasis, Disease, and Drug Intervention Nehemiah Zewde and Dimitrios Morikis* Department of Bioengineering University of California, Riverside *Corresponding author. E-mail address: [email protected] (D. Morikis) Key words: Computational ODE model; immune system regulation; complement system pathways; disease model; Factor H; compstatin; eculizumab. Page 1 of 33 bioRxiv preprint doi: https://doi.org/10.1101/225029; this version posted November 25, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Highlights • Computational model describing dynamics of complement system activation pathways • Complement dysregulation leads to deviation from homeostasis and to inflammatory diseases • Model identifies biomarkers to quantify the effects of complement dysregulation • Known drugs restore impaired dynamics of complement biomarkers under dysregulation • Disease-specific models are suitable for diagnosis and patient-specific drug treatment Page 2 of 33 bioRxiv preprint doi: https://doi.org/10.1101/225029; this version posted November 25, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract The complement system is a part of innate immunity that rapidly removes invading pathogens and impaired host-cells. Activation of the complement system is balanced under homeostasis by regulators that protect healthy host-cells.