Coagulation Simplified…
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Factor XIII and Fibrin Clot Properties in Acute Venous Thromboembolism
International Journal of Molecular Sciences Review Factor XIII and Fibrin Clot Properties in Acute Venous Thromboembolism Michał Z ˛abczyk 1,2 , Joanna Natorska 1,2 and Anetta Undas 1,2,* 1 John Paul II Hospital, 31-202 Kraków, Poland; [email protected] (M.Z.); [email protected] (J.N.) 2 Institute of Cardiology, Jagiellonian University Medical College, 31-202 Kraków, Poland * Correspondence: [email protected]; Tel.: +48-12-614-30-04; Fax: +48-12-614-21-20 Abstract: Coagulation factor XIII (FXIII) is converted by thrombin into its active form, FXIIIa, which crosslinks fibrin fibers, rendering clots more stable and resistant to degradation. FXIII affects fibrin clot structure and function leading to a more prothrombotic phenotype with denser networks, characterizing patients at risk of venous thromboembolism (VTE). Mechanisms regulating FXIII activation and its impact on fibrin structure in patients with acute VTE encompassing pulmonary embolism (PE) or deep vein thrombosis (DVT) are poorly elucidated. Reduced circulating FXIII levels in acute PE were reported over 20 years ago. Similar observations indicating decreased FXIII plasma activity and antigen levels have been made in acute PE and DVT with their subsequent increase after several weeks since the index event. Plasma fibrin clot proteome analysis confirms that clot-bound FXIII amounts associated with plasma FXIII activity are decreased in acute VTE. Reduced FXIII activity has been associated with impaired clot permeability and hypofibrinolysis in acute PE. The current review presents available studies on the role of FXIII in the modulation of fibrin clot properties during acute PE or DVT and following these events. -

Guideline on Clinical Investigation of Recombinant and Human Plasma-Derived 9 Factor IX Products’ (EMA/CHMP/BPWP/144552/2009 Rev
1 15 November 2018 2 EMA/CHMP/BPWP/144552/2009 rev. 2 Corr. 1 3 Committee for medicinal products for human use (CHMP) 4 Guideline on clinical investigation of recombinant and 5 human plasma-derived factor IX products 6 Draft Draft Agreed by Blood Products Working Party (BPWP) August 2018 Adopted by Committee for Medicinal Products for Human Use (CHMP) 15 November 2018 Start of public consultation 3 December 2018 End of public consultation 30 June 2019 7 8 This guideline replaces ‘Guideline on clinical investigation of recombinant and human plasma-derived 9 factor IX products’ (EMA/CHMP/BPWP/144552/2009 Rev. 1, Corr. 1) 10 Comments should be provided using this template. The completed comments form should be sent to [email protected] 11 Keywords Recombinant factor IX, plasma-derived factor IX, efficacy, safety, immunogenicity, inhibitor, thrombogenicity, anaphylactic reactions, potency assays 30 Churchill Place ● Canary Wharf ● London E14 5EU ● United Kingdom Telephone +44 (0)20 3660 6000 Facsimile +44 (0)20 3660 5555 Send a question via our website www.ema.europa.eu/contact An agency of the European Union © European Medicines Agency, 2019. Reproduction is authorised provided the source is acknowledged. 12 Guideline on the clinical investigation of recombinant and 13 human plasma-derived factor IX products 14 Table of contents 15 Executive summary ..................................................................................... 4 16 1. Introduction (background) ..................................................................... -

MONONINE (“Difficulty ® Monoclonal Antibody Purified in Concentrating”; Subject Recovered)
CSL Behring IU/kg (n=38), 0.98 ± 0.45 K at doses >95-115 IU/kg (n=21), 0.70 ± 0.38 K at doses >115-135 IU/kg (n=2), 0.67 K at doses >135-155 IU/kg (n=1), and 0.73 ± 0.34 K at doses >155 IU/kg (n=5). Among the 36 subjects who received these high doses, only one (2.8%) Coagulation Factor IX (Human) reported an adverse experience with a possible relationship to MONONINE (“difficulty ® Monoclonal Antibody Purified in concentrating”; subject recovered). In no subjects were thrombo genic complications MONONINE observed or reported.4 only The manufacturing procedure for MONONINE includes multiple processing steps that DESCRIPTION have been designed to reduce the risk of virus transmission. Validation studies of the Coagulation Factor IX (Human), MONONINE® is a sterile, stable, lyophilized concentrate monoclonal antibody (MAb) immunoaffinity chromatography/chemical treatment step and of Factor IX prepared from pooled human plasma and is intended for use in therapy nanofiltration step used in the production of MONONINE doc ument the virus reduction of Factor IX deficiency, known as Hemophilia B or Christmas disease. MONONINE is capacity of the processes employed. These studies were conducted using the rel evant purified of extraneous plasma-derived proteins, including Factors II, VII and X, by use of enveloped and non-enveloped viruses. The results of these virus validation studies utilizing immunoaffinity chromatography. A murine monoclonal antibody to Factor IX is used as an a wide range of viruses with different physicochemical properties are summarized in Table affinity ligand to isolate Factor IX from the source material. -

The Rare Coagulation Disorders
Treatment OF HEMOPHILIA April 2006 · No. 39 THE RARE COAGULATION DISORDERS Paula HB Bolton-Maggs Department of Haematology Manchester Royal Infirmary Manchester, United Kingdom Published by the World Federation of Hemophilia (WFH) © World Federation of Hemophilia, 2006 The WFH encourages redistribution of its publications for educational purposes by not-for-profit hemophilia organizations. In order to obtain permission to reprint, redistribute, or translate this publication, please contact the Communications Department at the address below. This publication is accessible from the World Federation of Hemophilia’s web site at www.wfh.org. Additional copies are also available from the WFH at: World Federation of Hemophilia 1425 René Lévesque Boulevard West, Suite 1010 Montréal, Québec H3G 1T7 CANADA Tel. : (514) 875-7944 Fax : (514) 875-8916 E-mail: [email protected] Internet: www.wfh.org The Treatment of Hemophilia series is intended to provide general information on the treatment and management of hemophilia. The World Federation of Hemophilia does not engage in the practice of medicine and under no circumstances recommends particular treatment for specific individuals. Dose schedules and other treatment regimes are continually revised and new side effects recognized. WFH makes no representation, express or implied, that drug doses or other treatment recommendations in this publication are correct. For these reasons it is strongly recommended that individuals seek the advice of a medical adviser and/or to consult printed instructions provided by the pharmaceutical company before administering any of the drugs referred to in this monograph. Statements and opinions expressed here do not necessarily represent the opinions, policies, or recommendations of the World Federation of Hemophilia, its Executive Committee, or its staff. -

Section 8: Hematology CHAPTER 47: ANEMIA
Section 8: Hematology CHAPTER 47: ANEMIA Q.1. A 56-year-old man presents with symptoms of severe dyspnea on exertion and fatigue. His laboratory values are as follows: Hemoglobin 6.0 g/dL (normal: 12–15 g/dL) Hematocrit 18% (normal: 36%–46%) RBC count 2 million/L (normal: 4–5.2 million/L) Reticulocyte count 3% (normal: 0.5%–1.5%) Which of the following caused this man’s anemia? A. Decreased red cell production B. Increased red cell destruction C. Acute blood loss (hemorrhage) D. There is insufficient information to make a determination Answer: A. This man presents with anemia and an elevated reticulocyte count which seems to suggest a hemolytic process. His reticulocyte count, however, has not been corrected for the degree of anemia he displays. This can be done by calculating his corrected reticulocyte count ([3% × (18%/45%)] = 1.2%), which is less than 2 and thus suggestive of a hypoproliferative process (decreased red cell production). Q.2. A 25-year-old man with pancytopenia undergoes bone marrow aspiration and biopsy, which reveals profound hypocellularity and virtual absence of hematopoietic cells. Cytogenetic analysis of the bone marrow does not reveal any abnormalities. Despite red blood cell and platelet transfusions, his pancytopenia worsens. Histocompatibility testing of his only sister fails to reveal a match. What would be the most appropriate course of therapy? A. Antithymocyte globulin, cyclosporine, and prednisone B. Prednisone alone C. Supportive therapy with chronic blood and platelet transfusions only D. Methotrexate and prednisone E. Bone marrow transplant Answer: A. Although supportive care with transfusions is necessary for treating this patient with aplastic anemia, most cases are not self-limited. -
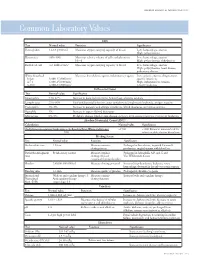
Common Laboratory Values
AmericAn AcAdemy of PediAtric dentistry Reference Manual 2006-2007 Resource Section 251 Common LaboratoryCommon Values Laboratory Values CBC Test Normal value Function Significance Hemoglobin 12-18 g/100 mL Measures oxygen carrying capacity of blood Low: hemorrhage, anemia High: polycythemia Hematocrit 35%-50% Measures relative volume of cells and plasma in Low: hemorrhage, anemia blood High: polycythemia, dehydration Red blood cell 4-6 million/mm3 Measures oxygen-carrying capacity of blood Low: hemorrhage, anemia High: polycythemia, heart disease, pulmonary disease White blood cell Measures host defense against inflammatory agents Low: aplastic anemia, drug toxicity, Infant 8,000-15,000/mm3 specific infections 4-7 y 6,000-15,000/mm3 High: inflammation, trauma, 8-18 y 4,500-13,500/mm3 toxicity, leukemia Differential Count Test Normal value Significance Neutrophils 54%-62% Increase in bacterial infections, hemorrhage, diabetic acidosis Lymphocytes 25%-30% Viral and bacterial infection, acute and chronic lymphocytic leukemia, antigen reaction Eosinophils 1%-3% Increase in parasitic and allergic conditions, blood dyscrasias, pernicious anemia Basophils 1% Increase in types of blood dyscrasias Monocytes 0%-9% Hodgkin’s disease, lipid storage disease, recovery from severe infections, monocytic leukemia Absolute Neutrophil Count (ANC) Calculation Normal value Significance (% Polymorphonuclear Leukocytes + % Bands)×Total White Cell Count >1500 <1000 Patient at increased risk for 100 infection; defer elective dental care Bleeding Screen Test -

Coagulation Testing: High Hematocrit-Anticoagulant Adjustment
PREANALYTIC PULSE Coagulation Testing: High Hematocrit-Anticoagulant Adjustment In 1980, CLSI released H21-A4 guidelines for coagulation testing, which included the recommendation to adjust/correct the amount of citrate in blue-top evacuated blood collection tubes for patients presenting hematocrits greater than 55%. In addition to the CLSI guidelines, the CAP hematology checklist HEM.22830 includes the question, “Are there documented guidelines for the detection and special handling of specimens with elevated hematocrits?” Principle The adjustment is to assure the plasma:anticoagulant ratio (not the blood:anticoagulant ratio) stays consistent. A patient with high hematocrit, greater than 55%, will result in less plasma after centrifugation. The plasma fraction will contain an increased concentration of sodium citrate anticoagulant. The increased concentration may result in falsely prolonged test results for Prothrombin Time (PT) and Activated Partial Thromboplastin Time (aPTT). Formula 1. To calculate the corrected 3.2% sodium citrated whole blood for hematocrit > 55%, adjust the citrate to the proper volume with the following formula: -3 C = (1.85 x 10 )(100-HCT)(Vblood) Where: C = volume of sodium citrate required for that volume of blood HCT = patient’s hematocrit V = volume of blood required in the blood collection tube (example if a 3mL tube is used the blood draw volume is 2.7mL) 1.85 x 10-3 is a constant (considering the citrate volume, blood volume, and citrate concentration). 2. Example: Patient has a Hct of 60% and the patient’s blood will be drawn into a 3mL VACUETTE® sodium citrate (blue-top) blood collection tube. Adjustment of the sodium citrate volume is calculated as: C = (1.85 x 10-3)(100-60)(2.7mL) C = 0.20mL (rounded up from 01.998mL) Remove: 0.30 - 0.20 = 0.10mL of sodium citrate to be removed (0.30 is the difference between the total tube volume of 3.0mL and the blood drawn into the tube of 2.7mL) Method The instructions to prepare an adjusted sodium citrate tube are to be documented in the Laboratory Standard Operating Procedures. -

Update on Antithrombin I (Fibrin)
©2007 Schattauer GmbH,Stuttgart AnniversaryIssueContribution Update on antithrombinI(fibrin) Michael W. Mosesson 1957–2007) The Blood Research Institute,BloodCenter of Wisconsin, Milwaukee,Wisconsin, USA y( Summary AntithrombinI(fibrin) is an important inhibitor of thrombin exosite 2.Thelatterreaction results in allostericchanges that generation that functions by sequestering thrombin in the form- down-regulate thrombin catalytic activity. AntithrombinIdefi- Anniversar ingfibrin clot,and also by reducing the catalytic activity of fibrin- ciency (afibrinogenemia), defectivethrombin binding to fibrin th boundthrombin.Thrombin binding to fibrin takesplace at two (antithrombin Idefect) found in certain dysfibrinogenemias (e.g. 50 classesofnon-substrate sites: 1) in thefibrin Edomain (two per fibrinogen Naples 1), or areduced plasma γ ’ chain content (re- molecule) throughinteractionwith thrombin exosite 1; 2) at a ducedantithrombin Iactivity),predispose to intravascular singlesite on each γ ’ chain through interaction with thrombin thrombosis. Keywords Fibrinogen,fibrin, thrombin, antithrombin I ThrombHaemost 2007; 98: 105–108 Introduction meric with respecttoits γ chains,and accounts for ~85% of human plasma fibrinogen. Thrombinbinds to its substrate, fibrinogen, through an anion- Low-affinity thrombin binding activity reflects thrombin ex- binding sitecommonlyreferred to as ‘exosite 1’ (1,2). Howell osite1bindinginEdomain of fibrin, as recentlydetailedbyana- recognized nearly acenturyago that the fibrin clot itself exhibits lysesofthrombin-fibrin -

Prolonged Partial Thromboplastin Time Without Bleeding History; Fletcher Factor Deficiency
Prolonged Partial Thromboplastin Time Without Bleeding History; Fletcher Factor Deficiency Celalettin ÜSTÜN*, Anand JILLELLA*, Linda HENDRIKS*, Mary JONAH**, Ferdane KUTLAR*, Russell BURGESS*, Abdullah KUTLAR* * Section of Hematology Oncology, Department of Medicine, Medical College of Georgia, ** Department of Pathology, Medical College of Georgia, Augusta, USA ABSTRACT A 67-year-old patient was admitted to the hospital to perform an esophagogastrectomy because a lesion at the lower esophagus was strongly suspicious for cancer. Her medical history and her family his- tory were negative for bleeding tendency or thrombosis. Her activated partial thromboplastin time (aPTT) was prolonged (44 s) whereas her prothrombin time (PT) was normal (11 s) presurgery. Mixing of her plasma with normal plasma corrected her prolonged aPTT (27.9 s). Prolonged incubation shortened the patient’s aPTT (36.3 s). Fletcher factor activity was found to be 50%. The patient underwent an esopha- gogastrectomy without bleeding complications under spinal anesthesia. Fletcher factor deficiency, a ra- re disorder, should be considered in patients who have no history of bleeding tendency with a prolonged aPTT. Surgical interventions are safe in these patients. Key Words: aPTT, Surgery, Fletcher factor, Prekallikrein. Turk J Haematol 2002;19(3): 417-419 Received: 20.06.2001 Accepted: 28.07.2001 INTRODUCTION unt[2]. Hematologists are frequently consulted for preoperative coagulation abnormalities. Preoperative evaluation of hemostasis is cru- cial to assess the risk of per-and peri-operative Prolonged aPTT is not an uncommon abnor- bleeding. The most effective screening method is mality encountered during preoperative evaluati- to obtain a thorough history of bleeding[1]. on. It may indicate the presence of antiphospholi- Preoperative screening tests mostly include acti- pid antibodies or a factor deficiency in the intrinsic vated partial thromboplastin time (aPTT), proth- and/or common pathways of blood coagulation. -

The Two Faces of Thrombosis: Coagulation Cascade and Platelet Aggregation. Are Platelets the Main Therapeutic Target
Thrombosis and Circulation Open Access Cimmino et al., J Thrombo Cir 2017, 3:1 Review Article Open Access The Two Faces of Thrombosis: Coagulation Cascade and Platelet Aggregation. Are Platelets the Main Therapeutic Target? Giovanni Cimmino*, Salvatore Fischetti and Paolo Golino Department of Cardio-Thoracic and Respiratory Sciences, Section of Cardiology, University of Campania “Luigi Vanvitelli”, Naples, Italy *Corresponding author: Giovanni Cimmino, MD, PhD, Department of Cardio-Thoracic and Respiratory Sciences, Section of Cardiology, University of Campania “Luigi Vanvitelli”, via Leonardo Bianchi, 180131 Naples, Italy. Tel: +39-081-7064175, Fax: +39-081-7064234; E-mail: [email protected] Received date: Dec 28, 2016, Accepted date: Jan 27, 2017, Published date: Jan 31, 2017 Copyright: © 2017 Giovanni C, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Abstract Acute thrombus formation is the pathophysiological substrate underlying several clinical conditions, such as acute coronary syndrome (ACS) and stroke. Activation of coagulation cascade is a key step of the thrombotic process: vessel injury results in exposure of the glycoprotein tissue factor (TF) to the flowing blood. Once exposed, TF binds factor VII/VIIa (FVII/FVIIa) and in presence of calcium ions, it forms a tertiary complex able to activate FX to FXa, FIX to FIXa, and FVIIa itself. The final step is thrombin formation at the site of vessel injury with subsequent platelet activation, fibrinogen to fibrin conversion and ultimately thrombus formation. Platelets are the key cells in primary hemostasis. -

Functional Plasminogen Activator Inhibitor 1 Is Retained on The
ARTICLE Hemostasis Functional plasminogen activator inhibitor 1 is Ferrata Storti Foundation retained on the activated platelet membrane following platelet activation Gael B. Morrow,° Claire S. Whyte and Nicola J. Mutch Institute of Medical Sciences, University of Aberdeen, Aberdeen, UK °Current address: Radcliffe Department of Medicine, University of Oxford, Oxford, UK Haematologica 2020 Volume 105(12):2824-2833 ABSTRACT latelets harbor the primary reservoir of circulating plasminogen acti- vator inhibitor 1 (PAI-1), but the reportedly low functional activity of Pthis pool of inhibitor has led to debate over its contribution to throm- bus stability. Here we analyze the fate of PAI-1 secreted from activated platelets and examine its role in maintaining thrombus integrity. Activation of platelets results in translocation of PAI-1 to the outer leaflet of the mem- brane, with maximal exposure in response to strong dual agonist stimula- tion. PAI-1 is found to co-localize in the 'cap' of phosphatidylserine-expos- ing platelets with its co-factor, vitronectin, and fibrinogen. Inclusion of tirofiban or Gly-Pro-Arg-Pro significantly attenuated exposure of PAI-1, indicating a crucial role for integrin αIIbb3 and fibrin in delivery of PAI-1 to the activated membrane. Separation of platelets post stimulation into sol- uble and cellular components revealed the presence of PAI-1 antigen and activity in both fractions, with approximately 40% of total platelet-derived PAI-1 remaining associated with the cellular fraction. Using a variety of fib- rinolytic models, we found that platelets produce a strong stabilizing effect against tissue plasminogen activator (tPA)-mediated clot lysis. Platelet lysate, as well as soluble and cellular fractions, stabilize thrombi against premature degradation in a PAI-1-dependent manner. -

We Get Biased on Things That Make Sense
DIALOGUE AND DISCUSSION We Get Biased on Things That Make Sense MERIH T. TESFAZGHI When I was teaching third year medical students, and bench- reaction might be characterized by a marked left shift lecturing clinical laboratory science students, I emphasized which may display a range of immature cells that are also the importance of clinical details and the history of patients seen in CML. Severe left shift especially in the absence of in laboratory test processing, especially in the evaluation of evident infection (or other underlying diseases) may peripheral blood films. During those times, one of my strongly suggest direct bone marrow involvement. In students stated, “but including details of patients might such scenario, clinical details of patients such as absence somehow affect the decision of the reviewer, and may lead to or presence of enlargement of extramedullar organs is Downloaded from a bias.” I responded that we are biased on things that make highly sought. sense. This means that we decide based on the evidence we see from a microscopy evaluation in the light of the clinical Ø The coexistence of multiple conditions in a given detail and history. How do clinical details and history help us patient. One condition may mask the presence of the reach decisions? other condition. For example, the coexistence of megaloblastic anemia with iron deficiency anemia. http://hwmaint.clsjournal.ascls.org/ If specimens are the “in vitro ambassadors” of patients to the Usually, megaloblastic anemia is characterized by laboratory, then properly completed request forms are their increased size of red blood cells (MCV), but when “credentials”.