Structural and Molecular Basis for Multifunctional Roles of Aminopeptidase N
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Enzymatic Encoding Methods for Efficient Synthesis Of
(19) TZZ__T (11) EP 1 957 644 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.: of the grant of the patent: C12N 15/10 (2006.01) C12Q 1/68 (2006.01) 01.12.2010 Bulletin 2010/48 C40B 40/06 (2006.01) C40B 50/06 (2006.01) (21) Application number: 06818144.5 (86) International application number: PCT/DK2006/000685 (22) Date of filing: 01.12.2006 (87) International publication number: WO 2007/062664 (07.06.2007 Gazette 2007/23) (54) ENZYMATIC ENCODING METHODS FOR EFFICIENT SYNTHESIS OF LARGE LIBRARIES ENZYMVERMITTELNDE KODIERUNGSMETHODEN FÜR EINE EFFIZIENTE SYNTHESE VON GROSSEN BIBLIOTHEKEN PROCEDES DE CODAGE ENZYMATIQUE DESTINES A LA SYNTHESE EFFICACE DE BIBLIOTHEQUES IMPORTANTES (84) Designated Contracting States: • GOLDBECH, Anne AT BE BG CH CY CZ DE DK EE ES FI FR GB GR DK-2200 Copenhagen N (DK) HU IE IS IT LI LT LU LV MC NL PL PT RO SE SI • DE LEON, Daen SK TR DK-2300 Copenhagen S (DK) Designated Extension States: • KALDOR, Ditte Kievsmose AL BA HR MK RS DK-2880 Bagsvaerd (DK) • SLØK, Frank Abilgaard (30) Priority: 01.12.2005 DK 200501704 DK-3450 Allerød (DK) 02.12.2005 US 741490 P • HUSEMOEN, Birgitte Nystrup DK-2500 Valby (DK) (43) Date of publication of application: • DOLBERG, Johannes 20.08.2008 Bulletin 2008/34 DK-1674 Copenhagen V (DK) • JENSEN, Kim Birkebæk (73) Proprietor: Nuevolution A/S DK-2610 Rødovre (DK) 2100 Copenhagen 0 (DK) • PETERSEN, Lene DK-2100 Copenhagen Ø (DK) (72) Inventors: • NØRREGAARD-MADSEN, Mads • FRANCH, Thomas DK-3460 Birkerød (DK) DK-3070 Snekkersten (DK) • GODSKESEN, -

PROTEOMIC ANALYSIS of HUMAN URINARY EXOSOMES. Patricia
ABSTRACT Title of Document: PROTEOMIC ANALYSIS OF HUMAN URINARY EXOSOMES. Patricia Amalia Gonzales Mancilla, Ph.D., 2009 Directed By: Associate Professor Nam Sun Wang, Department of Chemical and Biomolecular Engineering Exosomes originate as the internal vesicles of multivesicular bodies (MVBs) in cells. These small vesicles (40-100 nm) have been shown to be secreted by most cell types throughout the body. In the kidney, urinary exosomes are released to the urine by fusion of the outer membrane of the MVBs with the apical plasma membrane of renal tubular epithelia. Exosomes contain apical membrane and cytosolic proteins and can be isolated using differential centrifugation. The analysis of urinary exosomes provides a non- invasive means of acquiring information about the physiological or pathophysiological state of renal cells. The overall objective of this research was to develop methods and knowledge infrastructure for urinary proteomics. We proposed to conduct a proteomic analysis of human urinary exosomes. The first objective was to profile the proteome of human urinary exosomes using liquid chromatography-tandem spectrometry (LC- MS/MS) and specialized software for identification of peptide sequences from fragmentation spectra. We unambiguously identified 1132 proteins. In addition, the phosphoproteome of human urinary exosomes was profiled using the neutral loss scanning acquisition mode of LC-MS/MS. The phosphoproteomic profiling identified 19 phosphorylation sites corresponding to 14 phosphoproteins. The second objective was to analyze urinary exosomes samples isolated from patients with genetic mutations. Polyclonal antibodies were generated to recognize epitopes on the gene products of these genetic mutations, NKCC2 and MRP4. The potential usefulness of urinary exosome analysis was demonstrated using the well-defined renal tubulopathy, Bartter syndrome type I and using the single nucleotide polymorphism in the ABCC4 gene. -

Cloud-Clone 16-17
Cloud-Clone - 2016-17 Catalog Description Pack Size Supplier Rupee(RS) ACB028Hu CLIA Kit for Anti-Albumin Antibody (AAA) 96T Cloud-Clone 74750 AEA044Hu ELISA Kit for Anti-Growth Hormone Antibody (Anti-GHAb) 96T Cloud-Clone 74750 AEA255Hu ELISA Kit for Anti-Apolipoprotein Antibodies (AAHA) 96T Cloud-Clone 74750 AEA417Hu ELISA Kit for Anti-Proteolipid Protein 1, Myelin Antibody (Anti-PLP1) 96T Cloud-Clone 74750 AEA421Hu ELISA Kit for Anti-Myelin Oligodendrocyte Glycoprotein Antibody (Anti- 96T Cloud-Clone 74750 MOG) AEA465Hu ELISA Kit for Anti-Sperm Antibody (AsAb) 96T Cloud-Clone 74750 AEA539Hu ELISA Kit for Anti-Myelin Basic Protein Antibody (Anti-MBP) 96T Cloud-Clone 71250 AEA546Hu ELISA Kit for Anti-IgA Antibody 96T Cloud-Clone 71250 AEA601Hu ELISA Kit for Anti-Myeloperoxidase Antibody (Anti-MPO) 96T Cloud-Clone 71250 AEA747Hu ELISA Kit for Anti-Complement 1q Antibody (Anti-C1q) 96T Cloud-Clone 74750 AEA821Hu ELISA Kit for Anti-C Reactive Protein Antibody (Anti-CRP) 96T Cloud-Clone 74750 AEA895Hu ELISA Kit for Anti-Insulin Receptor Antibody (AIRA) 96T Cloud-Clone 74750 AEB028Hu ELISA Kit for Anti-Albumin Antibody (AAA) 96T Cloud-Clone 71250 AEB264Hu ELISA Kit for Insulin Autoantibody (IAA) 96T Cloud-Clone 74750 AEB480Hu ELISA Kit for Anti-Mannose Binding Lectin Antibody (Anti-MBL) 96T Cloud-Clone 88575 AED245Hu ELISA Kit for Anti-Glutamic Acid Decarboxylase Antibodies (Anti-GAD) 96T Cloud-Clone 71250 AEK505Hu ELISA Kit for Anti-Heparin/Platelet Factor 4 Antibodies (Anti-HPF4) 96T Cloud-Clone 71250 CCA005Hu CLIA Kit for Angiotensin II -

Characterization of Aminopeptidase in the Free-Living Nematode Panagrellus Redivivus: Subcellular Distribution and Possible Role in Neuropeptide Metabolism E
Journal of Nematology 39(2):153–160. 2007. © The Society of Nematologists 2007. Characterization of Aminopeptidase in the Free-living Nematode Panagrellus redivivus: Subcellular Distribution and Possible Role in Neuropeptide Metabolism E. P. Masler Abstract: Aminopeptidase was detected in homogenates of the free-living nematode Panagrellus redivivus with the aminoacyl substrate L-alanine-4-nitroanilide. Subcellular distribution of activity was 80% soluble and 20% membrane-associated. Aminopep- tidases in the two fractions differed in affinity for Ala-4-NA, with Km’s of 0.65 mM (soluble) and 2.90 mM (membrane). Specific activities (units/mg) at pH 7.8, 27°C were 9.10 (soluble) and 14.30 (membrane). Each enzyme was competitively inhibited by amastatin (90% at 100 µM inhibitor, IC50 = 3.7 µM) and inhibited by puromycin (30% at 500 µM) and 1,10-phenanthroline (IC50’s:; 148 µM, soluble; 89 µM, membrane). Activity was restored by Zn++, with maximum recoveries of 50% (soluble) and 90% (mem- ∼ brane), each at 23 µM ZnCl2. Estimated molecular masses for each were 150 kDa. FMRFamide-like neuropeptides behaved as competitive inhibitors. Modification of the N-terminal F of FMRFamide weakened inhibition by 95%, suggesting that the N-terminus is essential for binding to the enzyme. Two nematode FMRFamides, APKPFIRFa and RNKFEFIRFa, were the most potent tested. This is the first biochemical characterization of aminopeptidase in a free-living nematode other than Caenorhabditis elegans and demon- strates the high selectivity of the P. redivivus enzymes for neuropeptide substrates. Key words: FMRFamide-like peptide, inhibitor; membrane, metallopeptidase, neuropeptide, protease Nematodes, like other eukaryotic organisms, depend reproduction (Day and Maule, 1999; Maule et al., 2002; upon proteolytic enzymes for the regulation of essential Rogers et al., 2003). -

Code Lists Page 1
Code lists Page 1 Code lists AESEV Page 2 AESEV Codelist Name: Severity/Intensity Scale for Adverse Events Description: A scale that defines the degree or state of disease existing in a patient as a result of the occurrence of an adverse event. (NCI) C41338,1; Grade 1 C41339,2; Grade 2 C41340,3; Grade 3 AGEU Page 3 AGEU Codelist Name: Age Unit Description: Those units of time that are routinely used to express the age of a subject. C25301,Days C25529,Hours; h; hr C29846,Month C29844,Week C29848,Year CMDOSFRM Page 4 CMDOSFRM Codelist Name: Concomitant Medication Dose Form Description: A terminology subset of the CDISC SDTM Pharmaceutical Dosage Form codelist created for CDASH Concomitant Medication Dose Form codelist. (NCI) C42887,AEROSOL; aer C25158,CAPSULE; cap C28944,CREAM; C42933,GAS; C42934,GEL; C42966,OINTMENT; oint C42968,PATCH; C42972,POWDER; C42989,SPRAY; C42993,SUPPOSITORY; supp C42994,SUSPENSION; susp C42998,TABLET; tab CMDOSFRQ Page 5 CMDOSFRQ Codelist Name: Concomitant Medication Dosing Frequency per Interval Description: A terminology subset of the CDISC SDTM Frequency codelist created for CDASH Concomitant Medication Dosing Frequency per Interval codelist. (NCI) C64496,BID; BD; Twice per day C64499,PRN; As needed C25473,QD; Daily C64530,QID; 4 times per day C64498,QM; Every Month; Per Month C64525,QOD; Every other day C64527,TID; 3 times per day C17998,UNKNOWN; U; UNK; Unknown CMDOSU Page 6 CMDOSU Codelist Name: Concomitant Medication Dose Units Description: A terminology subset of the CDISC SDTM Unit codelist created for CDASH Concomitant Medication Dose Units codelist. (NCI) C48480,CAPSULE; Capsule Dosing Unit; cap C48155,g; Gram C48579,IU; IE; International Unit C28253,mg; Milligram C28254,mL; Milliliter; cm3 C65060,PUFF; Puff Dosing Unit C48542,TABLET; Tablet Dosing Unit; tab C48152,ug; Microgram; mcg CMROUTE Page 7 CMROUTE Codelist Name: Concomitant Medication Route of Administration Description: A terminology subset of the CDISC SDTM Route codelist created for CDASH Concomitant Medication Route of Administration codelist. -
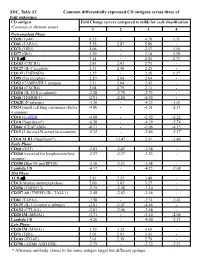
SDC, Table S1. Common Differentially Expressed CD Antigens
SDC, Table S1. Common differentially expressed CD antigens across three of four outcomes CD antigen Fold Change (severe compared to mild) for each classification (Common or alternate name) 1 2 3 4 Pretransplant Phase CD28 (Tp44) 6.32 - 4.79 3.71 CD81 (TAPA1) 5.58 2.87 5.86 - CD71 (TfR1) 4.06 - 2.57 3.16 CD77 (Gb3) 3.29 - 3.18 3.29 TCR ab 3.14 - 2.01 2.73 CD183 (CXCR3) 2.43 2.93 2.71 - CD127 (IL-7 receptor) 2.31 2.48 2.25 - CD137 (TNFRSF9) 2.27 - 2.35 2.27 CD95 (Fas receptor) 2.20 2.04 2.64 - CD52 (CAMPATH-1 antigen) 2.11 2.04 2.43 - CD184 (CXCR4) 2.04 2.79 2.11 - CD210 (IL-10 R a subunit) -2.28 -2.79 -2.73 - CD40 (TNFRSF5) -2.91 -2.20 -4.92 - CD62E (E-selectin) -3.36 - -4.23 -3.01 CD24 (small cell lung carcinoma cluster -4.00 - -3.51 -2.33 4 antigen) CD16 (FcγRIII) -4.08 - -2.92 -2.22 CD10 (Neprilysin*) -6.28 - -4.29 -5.74 CD66c (CEACAM6) -8.51 - -5.06 -6.15 CD15 (3-fucosyl-N-acetyl-lactosamine) -9.32 - -5.86 -5.17 CD10 ALB1 (Neprilysin*) - 12.47 2.51 -3.46 Early Phase CD60 (GD3) -2.03 -2.43 -2.58 - CD260 (reserved for lymphotoxin beta -2.57 -2.27 -3.23 - receptor) CD180 (Bgp-95 and RP105) -3.10 -2.33 -3.48 - Lambda 1/8 -4.17 - -4.23 -2.68 Mid Phase TCR ab 2.81 2.22 3.48 - CD13(Alanine aminopeptidase) 2.00 3.03 3.27 - CD256 (TNFSF13) -2.19 -2.48 -3.10 - CD257 AS (TNFSF13B / TALL1) -2.48 -2.62 -3.46 - CD81 (TAPA1) -2.58 - -2.31 -2.43 CD125 (IL-5 receptor a subunit) -2.81 -3.07 -4.66 - CD152 (CTLA-4) -2.81 -2.06 -3.48 - CD20 IM (MS4A1) -3.71 - -3.10 -2.04 Lambda 1/8 -4.26 - -4.00 -2.13 Late Phase CD20 IM (MS4A1) 3.39 3.32 4.69 - CD71 (TfR1) 2.03 2.93 3.01 - CD103 (ITGAE) 2.03 2.39 2.79 - CD79b (AGM6 AND IGB) -2.79 - -2.32 -2.23 * Alternate antibody clones to the same antigen target but different epitope. -

Aminopeptidases in Cardiovascular and Renal Function. Role As Predictive Renal Injury Biomarkers
International Journal of Molecular Sciences Review Aminopeptidases in Cardiovascular and Renal Function. Role as Predictive Renal Injury Biomarkers Félix Vargas 1, Rosemary Wangesteen 2, Isabel Rodríguez-Gómez 1 and Joaquín García-Estañ 3,* 1 Depto. Fisiologia, Fac. Medicina, Universidad de Granada, 18071 Granada, Spain; [email protected] (F.V.); [email protected] (I.R.-G.) 2 Depto. Ciencias de la salud, Universidad de Jaén, 23071 Jaén, Spain; [email protected] 3 Depto. Fisiologia, Fac. Medicina, IMIB, Universidad de Murcia, 30120 Murcia, Spain * Correspondence: [email protected] Received: 23 June 2020; Accepted: 3 August 2020; Published: 5 August 2020 Abstract: Aminopeptidases (APs) are metalloenzymes that hydrolyze peptides and polypeptides by scission of the N-terminus amino acid and that also participate in the intracellular final digestion of proteins. APs play an important role in protein maturation, signal transduction, and cell-cycle control, among other processes. These enzymes are especially relevant in the control of cardiovascular and renal functions. APs participate in the regulation of the systemic and local renin–angiotensin system and also modulate the activity of neuropeptides, kinins, immunomodulatory peptides, and cytokines, even contributing to cholesterol uptake and angiogenesis. This review focuses on the role of four key APs, aspartyl-, alanyl-, glutamyl-, and leucyl-cystinyl-aminopeptidases, in the control of blood pressure (BP) and renal function and on their association with different cardiovascular and renal diseases. In this context, the effects of AP inhibitors are analyzed as therapeutic tools for BP control and renal diseases. Their role as urinary biomarkers of renal injury is also explored. The enzymatic activities of urinary APs, which act as hydrolyzing peptides on the luminal surface of the renal tubule, have emerged as early predictive renal injury biomarkers in both acute and chronic renal nephropathies, including those induced by nephrotoxic agents, obesity, hypertension, or diabetes. -

(12) Patent Application Publication (10) Pub. No.: US 2004/0081648A1 Afeyan Et Al
US 2004.008 1648A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2004/0081648A1 Afeyan et al. (43) Pub. Date: Apr. 29, 2004 (54) ADZYMES AND USES THEREOF Publication Classification (76) Inventors: Noubar B. Afeyan, Lexington, MA (51) Int. Cl." ............................. A61K 38/48; C12N 9/64 (US); Frank D. Lee, Chestnut Hill, MA (52) U.S. Cl. ......................................... 424/94.63; 435/226 (US); Gordon G. Wong, Brookline, MA (US); Ruchira Das Gupta, Auburndale, MA (US); Brian Baynes, (57) ABSTRACT Somerville, MA (US) Disclosed is a family of novel protein constructs, useful as Correspondence Address: drugs and for other purposes, termed “adzymes, comprising ROPES & GRAY LLP an address moiety and a catalytic domain. In Some types of disclosed adzymes, the address binds with a binding site on ONE INTERNATIONAL PLACE or in functional proximity to a targeted biomolecule, e.g., an BOSTON, MA 02110-2624 (US) extracellular targeted biomolecule, and is disposed adjacent (21) Appl. No.: 10/650,592 the catalytic domain So that its affinity Serves to confer a new Specificity to the catalytic domain by increasing the effective (22) Filed: Aug. 27, 2003 local concentration of the target in the vicinity of the catalytic domain. The present invention also provides phar Related U.S. Application Data maceutical compositions comprising these adzymes, meth ods of making adzymes, DNA's encoding adzymes or parts (60) Provisional application No. 60/406,517, filed on Aug. thereof, and methods of using adzymes, Such as for treating 27, 2002. Provisional application No. 60/423,754, human Subjects Suffering from a disease, Such as a disease filed on Nov. -

Human Aminopeptidases: a Review of the Literature
Sanderink et al: Aminopeptidases, a review 795 J. Clin. Chem. Clin. Biochem. Vol. 26, 1988, pp. 795-807 © 1988 Waiter de Gruyter & Co. Berlin · New York Human Aminopeptidases: A Review of the Literature By G.-J. Sanderink, Y. Artur and G. Siesf Laboratoire du Centre de Medecine Preventive, UA CNRS n° 597, Vandoeuvre-les-Nancy, France (Received April 5/August 1, 1988) Summary: The aminopeptidases constitute a group of enzymes with closely related activities. In clinical chemistry the analysis of the aminopeptidases and of their multiple forms in serum has for a long time been hindered by considerable confusion concerning their identification, and by a lack of characterization. This is in part due to the often large, and sometimes overlapping substrate specificities of the aminopeptidases. This paper reviews the biochemical properties of the different aminopeptidases, the specificities of the assays used for their analysis in serum, some aspects of their multiple forms — which are especially known to occur for alanine äminopeptidase (EC 3.4.11.2) — and the importance of the determination of aminopeptidases and their multiple forms in clinical chemistry. Introduction ificities, pH optima, activators, etc. A list of some Aminopeptidases are enzymes which hydrolyse pep- human aminopeptidases acting on polypeptides is tide bonds near the N-terminal end of polypeptides. given in table 1. They can be subdivided into aminopeptidases which — substrate specificities; the different aminopepti- hydrolyse the first peptide bond (aminoacyl-peptide dases have a closely related enzymatic activity with hydrolases and iminoacyl-peptide hydrolases) and sometimes broad specificities. These specificities those which remove dipeptides from polypeptide overlap, so that many natural and synthetic sub- chains (dipeptidyl-peptide hydrolases). -

12) United States Patent (10
US007635572B2 (12) UnitedO States Patent (10) Patent No.: US 7,635,572 B2 Zhou et al. (45) Date of Patent: Dec. 22, 2009 (54) METHODS FOR CONDUCTING ASSAYS FOR 5,506,121 A 4/1996 Skerra et al. ENZYME ACTIVITY ON PROTEIN 5,510,270 A 4/1996 Fodor et al. MICROARRAYS 5,512,492 A 4/1996 Herron et al. 5,516,635 A 5/1996 Ekins et al. (75) Inventors: Fang X. Zhou, New Haven, CT (US); 5,532,128 A 7/1996 Eggers Barry Schweitzer, Cheshire, CT (US) 5,538,897 A 7/1996 Yates, III et al. s s 5,541,070 A 7/1996 Kauvar (73) Assignee: Life Technologies Corporation, .. S.E. al Carlsbad, CA (US) 5,585,069 A 12/1996 Zanzucchi et al. 5,585,639 A 12/1996 Dorsel et al. (*) Notice: Subject to any disclaimer, the term of this 5,593,838 A 1/1997 Zanzucchi et al. patent is extended or adjusted under 35 5,605,662 A 2f1997 Heller et al. U.S.C. 154(b) by 0 days. 5,620,850 A 4/1997 Bamdad et al. 5,624,711 A 4/1997 Sundberg et al. (21) Appl. No.: 10/865,431 5,627,369 A 5/1997 Vestal et al. 5,629,213 A 5/1997 Kornguth et al. (22) Filed: Jun. 9, 2004 (Continued) (65) Prior Publication Data FOREIGN PATENT DOCUMENTS US 2005/O118665 A1 Jun. 2, 2005 EP 596421 10, 1993 EP 0619321 12/1994 (51) Int. Cl. EP O664452 7, 1995 CI2O 1/50 (2006.01) EP O818467 1, 1998 (52) U.S. -

Purification, Characterization, and N-Terminal Amino Acid Sequence
J. Biochem. 124, 428-433 (1998) Dipeptidyl Peptidase IV from Human Serum: Purification, Characterization, and N-Terminal Amino Acid Sequence Sachiko Iwaki-Eaawa.*"' Yasuhiro Watanabe,* Yasuoki Kikuya,* and Yukio Fujimoto•õ *Department of Clinical Biochemistry and 'Department of Biochemistry , Hokkaido College of Pharmacy, 7-1 Katsuraoka-cho, Otaru, Hokkaido 047-0264 Received for publication, March 23, 1998 Dipeptidyl peptidase IV (DPP IV) in normal human serum was purified 14,400-fold with a 25% yield to homogeneity. The molecular weight of the purified enzyme was approximately 110,000 on SDS-PAGE, almost the same as that of human kidney membrane-bound DPP IV. No difference was found between the two enzymes enzymologically and immunologically, either in substrate specificity, susceptibility to inhibitors, or cross-reactivity with an anti- rat kidney DPP IV antibody, or in their ability to bind adenosine deaminase. However, the N-terminal amino acid sequence of serum DPP IV lacked the transmembrane domain of the membrane-bound enzyme and started at the 39th position, serine, from the N-terminus predicted from the cDNA nucleotide sequence. These results suggest that membrane-bound DPP IV loses its transmembrane domain upon release into the serum, and that its structure on the plasma membrane is not required for its binding to adenosine deaminase. Key words: adenosine deaminase, dipeptidyl peptidase IV, human serum, kidney, N-termi- nal amino acid sequence. In recent years, special interest has been paid to the with the susceptibility of a T cell line to infection with aminopeptidases localized on the plasma membrane of monocytotropic HIV-1 (19). Oravecz, T. -

Development of a Primer System to Study Abundance and Diversity of the Gene Coding for Alanine Aminopeptidase Pepn Gene in Gram-Negative Soil Bacteria
Journal of Microbiological Methods 91 (2012) 14–21 Contents lists available at SciVerse ScienceDirect Journal of Microbiological Methods journal homepage: www.elsevier.com/locate/jmicmeth Development of a primer system to study abundance and diversity of the gene coding for alanine aminopeptidase pepN gene in Gram-negative soil bacteria Esther Enowashu a, Ellen Kandeler a, Michael Schloter b,⁎, Frank Rasche c, Marion Engel b a Institute of Soil Science and Land Evaluation, Soil Biology Section, University of Hohenheim, Emil-Wolf Straße 27, Stuttgart, D-70593, Germany b Research Unit for Environmental Genomics, Helmholtz Zentrum München, National Research Centre for Environmental Health, Ingolstädter Landstraße1, Neuherberg, D-85764, Germany c Department of Plant Production in the Tropics and Subtropics, University of Hohenheim, Garbenstraße 13, Stuttgart, D-70593, Germany article info abstract Article history: A new set of primers was developed allowing the specific detection of the pepN gene (coding for alanine Received 11 June 2012 aminopeptidase) from Gram-negative bacteria. The primers were designed in silico by sequence alignments Accepted 3 July 2012 based on available DNA sequence data. The PCR assay was validated using DNA from selected pure cultures. Available online 14 July 2012 The analysis of gene libraries from extracted DNA from different soil samples revealed a high diversity of pepN related sequences mainly related to α-Proteobacteria. Most sequences obtained from clone libraries Keywords: were closely related to already