Monitoring Atom Transfer Radical Polymerisation Using 14C- Radiolabelled Initiators
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
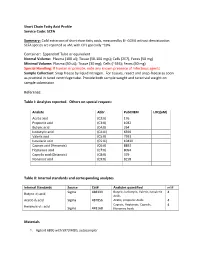
Short Chain Fatty Acid Profile Service Code: SCFA
Short Chain Fatty Acid Profile Service Code: SCFA Summary: Cold extraction of short chain fatty acids, measured by EI- GCMS without derivatization. SCFA species are reported as uM, with CV's generally ~10%. Container: Eppendorf Tube or equivalent Normal Volume: Plasma (100 ul); Tissue (50-100 mgs); Cells (2E7), Feces (50 mg) Minimal Volume: Plasma (50 uL); Tissue (30 mg); Cells (~5E6); Feces (40 mg) Special Handling: If human or primate, note any known presence of infectious agents. Sample Collection: Snap freeze by liquid nitrogen. For tissues, resect and snap-freeze as soon as practical in tared centrifuge tube. Provide both sample weight and tared vial weight on sample submission Reference: Table I: Analytes reported. Others on special request: Analyte Abbr. PubCHEM LOQ(uM) Acetic acid (C2:0) 176 Propionic acid (C3:0) 1032 Butyric acid (C4:0) 264 Isobutyric acid (C4:0i) 6590 Valeric acid (C5:0) 7991 Isovaleric acid (C5:0i) 10430 Caproic acid (Hexanoic) (C6:0) 8892 Heptanoic acid (C7:0) 8094 Caprylic acid (Octanoic) (C8:0) 379 Nonanoic acid (C9:0) 8158 Table II: Internal standards and corresponding analytes Internal Standards Source Cat# Analytes quantified mM Sigma 488399 Butyric, Isobutyric, Valeric, Isovaleric 4 Butyric-d7 acid Acids Acetic-d3 acid Sigma 487856 Acetic, propionic Acids 4 Caproic, Heptanoic, Caprylic, 4 Hexanoic-d11 acid Sigma 448168 Nonanoic Acids Materials 1. Agilent 6890 with 5973 MSD, autosampler 2. Vortexer 3. Refrigerated centrifuge, capable of 13,000g with eppendorf tube compatible rotor 4. ice bucket, ice 5. Balance 6. Prepared stock solutions of short chain fatty acid standards and isotope-labeled short chain fatty acid internal standards. -

Federal Register/Vol. 81, No. 250/Thursday, December 29, 2016
95886 Federal Register / Vol. 81, No. 250 / Thursday, December 29, 2016 / Rules and Regulations C. Regulatory Flexibility Act (RFA) I. National Technology Transfer and ENVIRONMENTAL PROTECTION Advancement Act (NTTAA) AGENCY This action is not subject to the RFA. This rulemaking does not involve The RFA applies only to rules subject to 40 CFR Part 180 notice-and-comment rulemaking technical standards. requirements under the APA, 5 U.S.C. [EPA–HQ–OPP–2016–0007 and EPA–HQ– J. Executive Order 12898: Federal OPP–2016–0008; FRL–9950–40] 553, or any other statute. This rule is not Actions To Address Environmental subject to notice-and-comment Justice in Minority Populations and Isobutyl Acetate and Isobutyric Acid; requirements because the agency has Low-Income Populations Exemption From the Requirement of a invoked the APA ‘‘good cause’’ The EPA believes that this action is Tolerance exemption under 5 U.S.C. 553(b). not subject to Executive Order 12898 (59 AGENCY: Environmental Protection FR 7629, February 16, 1994) because it D. Unfunded Mandates Reform Act Agency (EPA). (UMRA) does not establish an environmental health or safety standard. This good ACTION: Final rule. This action does not contain any cause final action simply extends the SUMMARY: This regulation establishes unfunded mandate of $100 million or date for the EPA to take action on a more as described in UMRA, 2 U.S.C. exemptions from the requirement of a petition and does not have any impact tolerance for residues of isobutyl acetate 1531–1538, and does not significantly or on human health or the environment. -
![[Agr. Biol. Chem., Vol. 36, No. 1, P. 112---119, 1972] Studies of the Peptide Antibiotic Suzukacillin Part II by Takaaki OOKA An](https://docslib.b-cdn.net/cover/9902/agr-biol-chem-vol-36-no-1-p-112-119-1972-studies-of-the-peptide-antibiotic-suzukacillin-part-ii-by-takaaki-ooka-an-399902.webp)
[Agr. Biol. Chem., Vol. 36, No. 1, P. 112---119, 1972] Studies of the Peptide Antibiotic Suzukacillin Part II by Takaaki OOKA An
[Agr. Biol. Chem., Vol. 36, No. 1, p. 112---119,1972] Studies of the Peptide Antibiotic Suzukacillin Part II By Takaaki OOKA and Isao TAKEDA TechnicalResearch Laboratory, Asahi ChemicalIndustry Co., Ltd., Nakadai-cho3-27, Itabashi-kuTokyo ReceivedJuly 26, 1971 Suzukacillin produced by Trichoderma viride 6301 strain was purified and shown to be separated into two components A and B on thin-layer chromatography. The component A was isolated and crystallized from the mixture of components by alumina column chro matography. The component A is composed of six amino acids, Gly, Glu, Ala, Pro, Val, Leu and an unknown amino acid. This unknown amino acid was identified as a-amino isobutyric acid. It is supposed that a-amino isobutyrie acid is biosynthesized mainly from L-valine by the isotopic experiments. Suzukacillin formation by Trichoderma viride 6301 was stimulated by the addition of L-Asn, GABA, L-Ser, Gly and L-Arg into the medium. Trichoderma viride 6301 strain producing Suzu ments were purchased as reagents from Tokyo Kasei kacillin was isolated by the author's laboratory. Co., Ltd. The following radioactive compounds were This organism accumulated noted amount of surplied by Dai-ichi Chemical Co., Ltd., with the in the peptide antibiotic. It was detected on dicated specific activities (ƒÊCi per mmole): Starch- TLC that the peptide antibiotic Suzukacillin (U)14C (30.8), pyruvic acid-(U)-14C (31.5), glycine- consists of two components A and B. A is (U)14C (45.2), L-alanine-(U)-14C (107), L-glutamic acid- (U)-14C (165), L-proline-(U)-14C (165), L-valine-(U)-14C a major and B is a minor component. -

Influence of Sargassum Horneri Mitigating Odorous Gas Emissions from Swine Manure Storage Facilities
sustainability Article Influence of Sargassum horneri Mitigating Odorous Gas Emissions from Swine Manure Storage Facilities Lavanya Madhavaraj 1, Ho-Dong Lim 1, Kong-Min Kim 1, Dae-Hyuk Kim 2 and Gui Hwan Han 1,* 1 Center for Industrialization of Agricultural and Livestock Microorganisms (CIALM), 241, Cheomdangwahag-ro, Jeongeup-si 56212, Korea; [email protected] (L.M.); [email protected] (H.-D.L.); [email protected] (K.-M.K.) 2 Department of Molecular Biology, Institute for Molecular Biology and Genetics, Jeonbuk National University, Jeonju 561–756, Korea; [email protected] * Correspondence: [email protected]; Tel.: +82-63-536-6713; Fax: +82-63-536-6003 Received: 4 August 2020; Accepted: 11 September 2020; Published: 15 September 2020 Abstract: Manures from livestock industries and farmyards should be managed for land application. Currently, a deep pit or barn system is adopted by many swine farms for manure management, therefore releasing harmful gases and rising the total global emissions of GHGs. This research focuses on the effectiveness of the brown seaweed Sargassum horneri as a masking agent to mitigate odor-generating gaseous pollutants and reduce the emissions of volatile fatty acids (VFAs) from swine manure storage facilities. Using an optimized procedure, we compared the gaseous emissions from two manure storage barns, one containing swine manure masked with S. horneri and the other without masking as a control, over a 30-day period. The results showed that, compared to the control, seaweed masking significantly reduced the sulfide and VFA contents. Furthermore, reductions of 99.48% in H S, 60 5.21% in NH and 74.28 2.14% in gaseous amine emissions were observed 2 ± 3 ± within the experimental period. -

Extraction of Benzoic Acid Dr
CHEM 2423 Extraction of Benzoic Acid Dr. Pahlavan EXPERIMENT 6 - Extraction Determination of Distribution Coefficient Purpose: a) To purify samples of organic compounds that are solids at room temperature b) To dissociate the impure sample in the minimum amount of an appropriate hot solvent Equipment / Materials: Benzoic acid solution ring& ring stand burette burette clamp spatula 0.1 M NaOH (or 0.02 M) solution separatory funnel methylene dichloride 125 ml Erlemeyer flask 10 and 50 mL graduated cylinders 50 and 100 mL beakers funnel Discussion: Crystallization, purification, and isolation (may only be restricted to a solid) are insufficient ways to separate mixtures of compounds. Extraction is the recovery of a substance from a mixture by bringing it into contact with a solvent, which dissolves the desired material. Partitioning is the separation between two distinct phases (immiscible liquids) and also called fractional separation. Like recrystallization and distillation, extraction is a separation technique frequently employed in the laboratory to isolate one or more components from a mixture. Unlike recrystallization and distillation, it does not yield a pure product; thus, the former techniques may be required to purify a product isolated by extraction. In the technical sense extraction is based on the principle of the equilibrium distribution of a substance (solute) between two immiscible phases, one of which is usually a solvent. The solvent need not be a pure liquid but may be a mixture of several solvents or a solution of some chemical reagent that will react with one or more components of the mixture being extracted to form a new substance soluble in the solution. -

Cigarette Additives, Carcinogens and Chemicals Nicotine
Cigarette Additives, Carcinogens and Chemicals Nicotine A Destructive Natural Pesticide Which ... Is extremely addictive when smoked Is extremely addictive when chewed Causes addiction as permanent as Is harder to quit than heroin or cocaine alcoholism Is not medicine and its use not therapy Is ineffective as a stand-alone quitting aid Prevents pre-cancerous cells from dying Accelerates cancer tumor growth rates Contributes to artery hardening Has a metabolite which may cause cancer May kill brain cells and impair memory Is linked to lung cancer Likely causes brain damage and Is also a fetus destroying teratogen depression Kills half of adult smokers 13-14 years Is beat by never taking another puff or early chew! 81 Cancer Causing Chemicals Have So Far Been Identified in Cigarettes Acetaldehyde Acetamide Acrylamide Acrylonitrile 2-Amino-3,4-dimethyl-3H-imidazo[4,5-f]quinoline (MeIQ) 3-Amino-1,4-dimethyl-5H-pyrido [4,3-b]indole (Trp-P-1) 2-Amino-l-methyl-6-phenyl-1H-imidazo [4,5-b]pyridine (PhlP) 2-Amino-6-methyldipyrido[1,2-a:3',2'-d]imidazole (Glu-P-1) 3-Amino-l-methyl-5H-pyrido {4,3-b]indole (Trp-P-2 2-Amino-3-methyl-9H-pyrido[2,3-b]indole (MeAaC) 2-Amino-9H-pyrido[2,3-b]indole (AaC) 4-Aminobiphenyl 2-Aminodipyrido[1,2-a:3',2'-d]imidazole (Glu-P-2) 0-Anisidine Arsenic Benz[a]anthracene Benzene Benzo[a]pyrene Benzo[b]fluoranthene Benzo[j]fluoranthene Benzo[k]fluoranthene Benzo[b]furan Beryllium 1,3-Butadiene Cadmium Catechol (1,2-benzenediol) p-Chloroaniline Chloroform Cobalt p,p'-DDT Dibenz[a,h]acridine Dibenz[a,j]acridine Dibenz(a,h)anthracene -

WIDESPREAD OCCURRENCE of SPECIFIC, HIGH AFFINITY BINDING SITES for AMINO ACIDS Stephen C
Vol. 80, No. 1, 1978 BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS January 13, 1978 Pages 161-168 WIDESPREAD OCCURRENCE OF SPECIFIC, HIGH AFFINITY BINDING SITES FOR AMINO ACIDS Stephen C. Bondy and Marilyn E. Harrington Departments of Neurology and Pharmacology University of Colorado Medical Center, 42DO East Ninth Avenue Denver, Colorado 80262 Received September 27, 1977 The high affinity binding of several amino acids to various membrane and protein preparations has been measured. Binding of radioactive amino acids suspected of being neurotransmitters and also of leucine and tyrosine to brain, liver and heart muscle membranes was saturable, reversible and stereospecific. Similar characteristics were found using chloroform-methanol extracted brain tissue and heat denatured albumin. Compounds thought to act as blockers of postsynaptic binding such as strychnine, bicuculline and kainic acid did not inhibit binding. Thus, specific high affinity interactions between amino acids and proteins are widespread and largely unrelated to neurotransmission. INTRODUCTION The affinity binding of several amino acids such as glutamic acid, glycine and gamma-aminobutyric acid has been reported in membrane preparations of brain (l-4). This binding has been used as a measure of postsynaptic sites specific for a particular amino acid neurotransmitter. Criteria used to establish the identity of these sites include: l) specificity of the interaction and also stereospecific selectivity in several cases; 2) saturable, reversible binding kinetics with a high affinity between membrane and ligand; 3) independence of binding on the presence of the sodium ion (5); 4) no energy dependence of the interaction, temperature should not have a major effect on binding velocity; 5) physiological relevance and appropriate inhibition by known antagonists. -

3.2.2 Table B: List of Dangerous Goods in Alphabetical Order the Following
Copyright © United Nations, 2008 3.2.2 Table B: List of dangerous goods in alphabetical order The following Table B is an alphabetical list of the substances and articles which are listed in the UN numerical order in Table A of 3.2.1. It does not form an integral part of ADN. It has been prepared, with all necessary care by the Secretariat of the United Nations Economic Commission for Europe, in order to facilitate the consultation of Annexes A and B, but it cannot be relied upon as a substitute for the careful study and observance of the actual provisions of those annexed Regulations which, in case of conflict, are deemed to be authoritative. NOTE 1: For the purpose of determining the alphabetical order the following information has been ignored, even when it forms part of the proper shipping name: numbers; Greek letters; the abbreviations "sec" and "tert"; and the letters "N" (nitrogen), "n" (normal), "o" (ortho) "m" (meta), "p" (para) and "N.O.S." (not otherwise specified). NOTE 2: The name of a substance or article in block capital letters indicates a proper shipping name (see 3.1.2). NOTE 3: The name of a substance or article in block capital letters followed by the word "see" indicates an alternative proper shipping name or part of a proper shipping name (except for PCBs) (see 3.1.2.1). NOTE 4: An entry in lower case letters followed by the word "see" indicates that the entry is not a proper shipping name; it is a synonym. NOTE 5: Where an entry is partly in block capital letters and partly in lower case letters, the latter part is considered not to be part of the proper shipping name (see 3.1.2.1). -

Drinking Water Advisory: Consumer Acceptability Advice and Health Effects Analysis on Methyl Tertiary-Butyl Ether (Mtbe) TABLE of CONTENTS
United States Office of Water EPA-822-F-97-009 Environmental Protection Agency 4304 December 1997 Drinking Water Advisory: Consumer Acceptability Advice and Health Effects Analysis on Methyl Tertiary-Butyl Ether (MtBE) TABLE OF CONTENTS LIST OF ABBREVIATIONS................................................................................ I FOREWORD......................................................................................... ii EXECUTIVE SUMMARY.................................................................... 1 1.0. INTRODUCTION...................................................................... 5 2.0. MtBE IN THE ENVIRONMENT............................................... 5 3.0. CHEMICAL AND PHYSICAL PROPERTIES........................... 6 4.0. TOXICOKINETICS.................................................................... 7 4.1. Dosimetry: Route-to-Route Extrapolation........................ 7 4.2. NSTC’s Extrapolation of Dose from Inhalation Exposure. 9 5.0. HEALTH EFFECTS DATA....................................................... 10 5.1. Human Studies................................................................ 10 5.2. Animal Studies................................................................ 11 5.2.1. Noncancer Effects............................................... 11 5.2.1.1. Acute and Subchronic........................... 11 5.2.1.2. Reproductive and Developmental Studies 13 Reproductive Studies............................ 13 Developmental Studies......................... 14 5.2.1.3. Neurotoxicity Studies.......................... -

Slr2 Displacements and Reductive Coupling of Ketones with Olefins in N,N-Diethylacetamide and N-Ethylpyrrolidone
Illeprinted lrom the.krurnalof Organic('hemistrl', ,14, 2;169 (1979).1 (-'opyrightO 19?9ltv the American (lhemical Society and reprinted bv permissiot-rol'the copyright owner. Slr2 Displacements and Reductive Coupling of Ketones with Olefins in N,N-Diethylacetamide and N-Ethylpyrrolidone Allan F. Sowinskiand GeorgeM. Whitesides* Departmentof Chemistrt',Massachusetts Institute of Technologt',Cambridge, Massachusetts 02139 ReceiuedMarch 8. 1979 N,l/-Diethylacetamide (DEA) and N-ethylpyrrolidone (NEP) are complementaryto hexamethylphosphoric triamide (HMPA), dimethylformamide,and dimethyl sulfoxideas solventsin which to carry out severalreactions commonly conductedin polar, aprotic media. Reactionof neopentyltosylates with lithium halidesin DEA and NEP givesgood yields of neopentylhalides. Ketonesand terminal olefinsare reductivelycoupled to tertiary, alcoholsin fair to good yields in mixtures containing NEP, sodium, and f erf -butyl alcohol;1-hepten-6-one and 1-octen-7-oneare cleanly cyclizedto five- and six-memberedrings, respectively,in good yield by this samemixture. Terminal olefins are reduced to alkanes in fair yield by DEA sodium ferf-butyl alcohol mixtures; di- and tetrasubstituted olefins are resistant to reduction bv this mixture. Hexamethylphosphorictriamide (HMPA) is a dipolar more limited value than HMPA for dissolving-metalre- aprotic solventr'2that is particularl5rvaluable for such duction of olefins. In addition, we describea useful new reactionsas SN2displacement with anionic nucleophilesrr'a reaction which occurs in NEP (but not in HMPA) bv and dissolving-metalreductions.s The routine use of which ketonesand terminal olefins are reductively coupled HMPA has beendiscouraged by the announcementthat to tertiary alcoholsby sodium-ferf-butyl alcohol mixtures. it may be carcinogenic.6In the courseof other studies, we required quantities of several neopentyl halides. Results and Discussion Compoundsof this type had proved in previouswork to Bimolecular Substitution in DEA and NEP. -

A Model Study on the Dynamics of the Amino Acid Content in Micrometeoroids During Atmospheric Entry
Article A Model Study on the Dynamics of the Amino Acid Content in Micrometeoroids during Atmospheric Entry Carlo Canepa Dipartimento di Chimica, Università di Torino, 10125 Torino, Italy; [email protected] Received: 22 August 2020; Accepted: 13 October 2020; Published: 30 October 2020 Abstract: A model for the survival of the amino acid content in micrometeorites and its possible transformations upon atmospheric entry is described. Since any organic species released in the atmosphere at high altitudes would exhibit a limited lifetime with respect to the typical timescale for atmospheric mixing (due to the substantial radiation field of the early Sun), only the organic content inside the meteorites could have contributed to prebiotic chemistry. It is thus important to determine both the amino acid loss from meteorites, due to both degassing and chemical degradation, and the extent of the chemical transformation of amino acids subjected to the increased temperature due to friction with the atmosphere. According to the interplay between the potential energy barrier and the pre-exponential factor in rate coefficients, either diffusion or chemical processing might be the dominant process following the rising temperature upon atmospheric entry. The possibility of the elimination of water from glycine to form aminoketene, or CO2 to form methylamine, ultimately driven by gravitational energy, was examined along with the more conventional formation of a peptide bond between two glycine units to afford Gly–Gly. While retention, degassing, and decarboxylation are the fastest processes, each dominating in different ranges of the initial velocity and radius, the formation of either Gly–Gly from bimolecular water elimination or aminoketene from unimolecular water elimination appears to be negligible. -

Chemical Compatibility Guide
20887Cvr:Layout 1 5/13/08 7:39 AM Page 2 Chemical Compatibility Guide Aro is pleased to present this selection guide to provide a convenient and informative reference point for pump selection. This information was compiled from information provided by material suppliers and manufacturers. The compatibility listings are intended as a guide only. We assume no liability for their accuracy on their use. The user should test under their own operating conditions to determine the suitability of any compound and material in a particular application. 20887Txt:43077 5/12/08 8:36 AM Page 1 20887Txt:43077 5/12/08 8:36 AM Page 1 THERMOPLASTIC ELASTOMERS (TPE’s) verses THERMOSETTING RUBBER DIAPHRAGMS Aro’s direction has been to move toward replacement of traditional thermoset rubber diaphragms with thermoplastic elastomers (TPE). Examples of TPE’s include: Santoprene, Nitrile (TPE) and Hytrel. TPE’s are manufactured using a plastic injection molding process where the resin, or diaphragm material, is melted and injected into a mold to produce the diaphragm. The advantages of this process includes: Features Benefits Diaphragm is molded to optimum shape Excellent flex life Homogenous part No delamination failures High performance resins Chemical, abrasion and flex life Injection molded/tight process control Low cost/consistent performance Laboratory testing has shown: Injection Mold Process Santoprene outperformed all rubber diaphragms except Buna in the mild abrasive fluids. Molded Diaphragm The Geolast diaphragm had equivalent life to the Buna diaphragms and was superior to the other rubbers compounds. 1 Teflon with the Santoprene backer exhibited the best flex life of all diaphragms during the test series.