United States Patent Office Patented Dec
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

2354 Metabolism and Ecology of Purine Alkaloids Ana Luisa Anaya 1
[Frontiers in Bioscience 11, 2354-2370, September 1, 2006] Metabolism and Ecology of Purine Alkaloids Ana Luisa Anaya 1, Rocio Cruz-Ortega 1 and George R. Waller 2 1Departamento de Ecologia Funcional, Instituto de Ecologia, Universidad Nacional Autonoma de Mexico. Mexico DF 04510, 2Department of Biochemistry and Molecular Biology, Oklahoma State University. Stillwater, OK 74078, USA TABLE OF CONTENTS 1. Abstract 2. Introduction 3. Classification of alkaloids 4. The importance of purine in natural compounds 5. Purine alkaloids 5.1. Distribution of purine alkaloids in plants 5.2. Metabolism of purine alkaloids 6. Biosynthesis of caffeine 6.1. Purine ring methylation 6.2. Cultured cells 7. Catabolism of caffeine 8. Caffeine-free and low caffeine varieties of coffee 8.1. Patents 9. Ecological role of alkaloids 9.1. Herbivory 9.2. Allelopathy 9.2.1. Mechanism of action of caffeine and other purine alkaloids in plants 10. Perspective 11. Acknowledgements 12. References 1. ABSTRACT 2. INTRODUCTION In this review, the biosynthesis, catabolism, Alkaloids are one of the most diverse groups of ecological significance, and modes of action of purine secondary metabolites found in living organisms. They alkaloids particularly, caffeine, theobromine and have many distinct types of structure, metabolic pathways, theophylline in plants are discussed. In the biosynthesis of and ecological and pharmacological activities. Many caffeine, progress has been made in enzymology, the amino alkaloids have been used in medicine for centuries, and acid sequence of the enzymes, and in the genes encoding some are still important drugs. Alkaloids have, therefore, N-methyltransferases. In addition, caffeine-deficient plants been prominent in many scientific fields for years, and have been produced. -
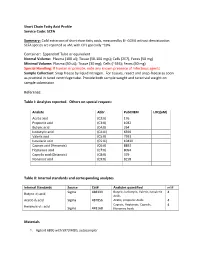
Short Chain Fatty Acid Profile Service Code: SCFA
Short Chain Fatty Acid Profile Service Code: SCFA Summary: Cold extraction of short chain fatty acids, measured by EI- GCMS without derivatization. SCFA species are reported as uM, with CV's generally ~10%. Container: Eppendorf Tube or equivalent Normal Volume: Plasma (100 ul); Tissue (50-100 mgs); Cells (2E7), Feces (50 mg) Minimal Volume: Plasma (50 uL); Tissue (30 mg); Cells (~5E6); Feces (40 mg) Special Handling: If human or primate, note any known presence of infectious agents. Sample Collection: Snap freeze by liquid nitrogen. For tissues, resect and snap-freeze as soon as practical in tared centrifuge tube. Provide both sample weight and tared vial weight on sample submission Reference: Table I: Analytes reported. Others on special request: Analyte Abbr. PubCHEM LOQ(uM) Acetic acid (C2:0) 176 Propionic acid (C3:0) 1032 Butyric acid (C4:0) 264 Isobutyric acid (C4:0i) 6590 Valeric acid (C5:0) 7991 Isovaleric acid (C5:0i) 10430 Caproic acid (Hexanoic) (C6:0) 8892 Heptanoic acid (C7:0) 8094 Caprylic acid (Octanoic) (C8:0) 379 Nonanoic acid (C9:0) 8158 Table II: Internal standards and corresponding analytes Internal Standards Source Cat# Analytes quantified mM Sigma 488399 Butyric, Isobutyric, Valeric, Isovaleric 4 Butyric-d7 acid Acids Acetic-d3 acid Sigma 487856 Acetic, propionic Acids 4 Caproic, Heptanoic, Caprylic, 4 Hexanoic-d11 acid Sigma 448168 Nonanoic Acids Materials 1. Agilent 6890 with 5973 MSD, autosampler 2. Vortexer 3. Refrigerated centrifuge, capable of 13,000g with eppendorf tube compatible rotor 4. ice bucket, ice 5. Balance 6. Prepared stock solutions of short chain fatty acid standards and isotope-labeled short chain fatty acid internal standards. -

Federal Register/Vol. 81, No. 250/Thursday, December 29, 2016
95886 Federal Register / Vol. 81, No. 250 / Thursday, December 29, 2016 / Rules and Regulations C. Regulatory Flexibility Act (RFA) I. National Technology Transfer and ENVIRONMENTAL PROTECTION Advancement Act (NTTAA) AGENCY This action is not subject to the RFA. This rulemaking does not involve The RFA applies only to rules subject to 40 CFR Part 180 notice-and-comment rulemaking technical standards. requirements under the APA, 5 U.S.C. [EPA–HQ–OPP–2016–0007 and EPA–HQ– J. Executive Order 12898: Federal OPP–2016–0008; FRL–9950–40] 553, or any other statute. This rule is not Actions To Address Environmental subject to notice-and-comment Justice in Minority Populations and Isobutyl Acetate and Isobutyric Acid; requirements because the agency has Low-Income Populations Exemption From the Requirement of a invoked the APA ‘‘good cause’’ The EPA believes that this action is Tolerance exemption under 5 U.S.C. 553(b). not subject to Executive Order 12898 (59 AGENCY: Environmental Protection FR 7629, February 16, 1994) because it D. Unfunded Mandates Reform Act Agency (EPA). (UMRA) does not establish an environmental health or safety standard. This good ACTION: Final rule. This action does not contain any cause final action simply extends the SUMMARY: This regulation establishes unfunded mandate of $100 million or date for the EPA to take action on a more as described in UMRA, 2 U.S.C. exemptions from the requirement of a petition and does not have any impact tolerance for residues of isobutyl acetate 1531–1538, and does not significantly or on human health or the environment. -
![[Agr. Biol. Chem., Vol. 36, No. 1, P. 112---119, 1972] Studies of the Peptide Antibiotic Suzukacillin Part II by Takaaki OOKA An](https://docslib.b-cdn.net/cover/9902/agr-biol-chem-vol-36-no-1-p-112-119-1972-studies-of-the-peptide-antibiotic-suzukacillin-part-ii-by-takaaki-ooka-an-399902.webp)
[Agr. Biol. Chem., Vol. 36, No. 1, P. 112---119, 1972] Studies of the Peptide Antibiotic Suzukacillin Part II by Takaaki OOKA An
[Agr. Biol. Chem., Vol. 36, No. 1, p. 112---119,1972] Studies of the Peptide Antibiotic Suzukacillin Part II By Takaaki OOKA and Isao TAKEDA TechnicalResearch Laboratory, Asahi ChemicalIndustry Co., Ltd., Nakadai-cho3-27, Itabashi-kuTokyo ReceivedJuly 26, 1971 Suzukacillin produced by Trichoderma viride 6301 strain was purified and shown to be separated into two components A and B on thin-layer chromatography. The component A was isolated and crystallized from the mixture of components by alumina column chro matography. The component A is composed of six amino acids, Gly, Glu, Ala, Pro, Val, Leu and an unknown amino acid. This unknown amino acid was identified as a-amino isobutyric acid. It is supposed that a-amino isobutyrie acid is biosynthesized mainly from L-valine by the isotopic experiments. Suzukacillin formation by Trichoderma viride 6301 was stimulated by the addition of L-Asn, GABA, L-Ser, Gly and L-Arg into the medium. Trichoderma viride 6301 strain producing Suzu ments were purchased as reagents from Tokyo Kasei kacillin was isolated by the author's laboratory. Co., Ltd. The following radioactive compounds were This organism accumulated noted amount of surplied by Dai-ichi Chemical Co., Ltd., with the in the peptide antibiotic. It was detected on dicated specific activities (ƒÊCi per mmole): Starch- TLC that the peptide antibiotic Suzukacillin (U)14C (30.8), pyruvic acid-(U)-14C (31.5), glycine- consists of two components A and B. A is (U)14C (45.2), L-alanine-(U)-14C (107), L-glutamic acid- (U)-14C (165), L-proline-(U)-14C (165), L-valine-(U)-14C a major and B is a minor component. -

Influence of Sargassum Horneri Mitigating Odorous Gas Emissions from Swine Manure Storage Facilities
sustainability Article Influence of Sargassum horneri Mitigating Odorous Gas Emissions from Swine Manure Storage Facilities Lavanya Madhavaraj 1, Ho-Dong Lim 1, Kong-Min Kim 1, Dae-Hyuk Kim 2 and Gui Hwan Han 1,* 1 Center for Industrialization of Agricultural and Livestock Microorganisms (CIALM), 241, Cheomdangwahag-ro, Jeongeup-si 56212, Korea; [email protected] (L.M.); [email protected] (H.-D.L.); [email protected] (K.-M.K.) 2 Department of Molecular Biology, Institute for Molecular Biology and Genetics, Jeonbuk National University, Jeonju 561–756, Korea; [email protected] * Correspondence: [email protected]; Tel.: +82-63-536-6713; Fax: +82-63-536-6003 Received: 4 August 2020; Accepted: 11 September 2020; Published: 15 September 2020 Abstract: Manures from livestock industries and farmyards should be managed for land application. Currently, a deep pit or barn system is adopted by many swine farms for manure management, therefore releasing harmful gases and rising the total global emissions of GHGs. This research focuses on the effectiveness of the brown seaweed Sargassum horneri as a masking agent to mitigate odor-generating gaseous pollutants and reduce the emissions of volatile fatty acids (VFAs) from swine manure storage facilities. Using an optimized procedure, we compared the gaseous emissions from two manure storage barns, one containing swine manure masked with S. horneri and the other without masking as a control, over a 30-day period. The results showed that, compared to the control, seaweed masking significantly reduced the sulfide and VFA contents. Furthermore, reductions of 99.48% in H S, 60 5.21% in NH and 74.28 2.14% in gaseous amine emissions were observed 2 ± 3 ± within the experimental period. -

Extraction of Benzoic Acid Dr
CHEM 2423 Extraction of Benzoic Acid Dr. Pahlavan EXPERIMENT 6 - Extraction Determination of Distribution Coefficient Purpose: a) To purify samples of organic compounds that are solids at room temperature b) To dissociate the impure sample in the minimum amount of an appropriate hot solvent Equipment / Materials: Benzoic acid solution ring& ring stand burette burette clamp spatula 0.1 M NaOH (or 0.02 M) solution separatory funnel methylene dichloride 125 ml Erlemeyer flask 10 and 50 mL graduated cylinders 50 and 100 mL beakers funnel Discussion: Crystallization, purification, and isolation (may only be restricted to a solid) are insufficient ways to separate mixtures of compounds. Extraction is the recovery of a substance from a mixture by bringing it into contact with a solvent, which dissolves the desired material. Partitioning is the separation between two distinct phases (immiscible liquids) and also called fractional separation. Like recrystallization and distillation, extraction is a separation technique frequently employed in the laboratory to isolate one or more components from a mixture. Unlike recrystallization and distillation, it does not yield a pure product; thus, the former techniques may be required to purify a product isolated by extraction. In the technical sense extraction is based on the principle of the equilibrium distribution of a substance (solute) between two immiscible phases, one of which is usually a solvent. The solvent need not be a pure liquid but may be a mixture of several solvents or a solution of some chemical reagent that will react with one or more components of the mixture being extracted to form a new substance soluble in the solution. -

Resolution of Racemic 5-Phenyl-2-Pentanol
Patentamt JEuropaischesEuropean Patent Office 11) Publication number: 0 078 145 Office europeen des brevets A1 © EUROPEAN PATENT APPLICATION © Int. ci * C 07 B 19/00 C 07 C 33/20, C 07 D 491/22 C 07 C 69/80 //C07D215/22, C07D221/12, (C07D491/22, 313/00, 221/00, 221/00, 209/00, 209/00) @ Priority: 22.10.81 US 313560 © Applicant: PFIZER INC. 235 East 42nd Street New York, N.Y.10017(US) © Date of publication of application: 04.05.83 Bulletin 83/18 © Inventor: Moore, Bernard Shields 39 Savi Avenue © Designated Contracting States: Waterford Connecticut(US) AT BE CH DE FR GB IT U LU NL SE © Representative: Graham, Philip Colin Christison et al, Pfizer Limited Ramsgate Road Sandwich, Kent CT13 9NJIGB) © Resolution of racemic 5-phenyl-2-pentanol. Racemic 5-phenyl-2-pentanol is resolved via its hemiph- thalate ester, followed by diastereomer salt formation with (+)-brucine, separation of the brucine salt of (S)-5-phenyl-2- pentylhemi-phthalate and recovery of the (S)-alcohol there- from. The (S)-5-phenyl-2-pentanol is a valuable intermediate for organic synthesis. The separated (+)-brucine salt of (S)-5-phenyl-2-pentylhemiphthalate is a novel and useful intermediate. This invention relates to a process for resolution of racemic 5-phenyl-2-pentanol to (S)-5-phenyl-2- pentanol, a valuable intermediate in the synthesis of analgesic agents. More specifically, the process comprises esterifying racemic 5-phenyl-2-pentanol to form the hemiphthalate ester, followed by treating said ester with (+)-brucine, separating (S)-5-phenyl- 2-pentylhemiphthalate(+)-brucine salt, decomposing said salt to regenerate (S)-5-phenyl-2-pentylhemiphthalate, and hydrolyzing said ester to (S)-5-phenyl-2-pentanol. -

Concentration and Frequency Dependent Dielectric Permittivity and Dielectric Loss Study of Brucine - Chloroform Solution Using Time Domain Reflectometry (TDR)
International Journal of Science and Research (IJSR) ISSN: 2319-7064 ResearchGate Impact Factor (2018): 0.28 | SJIF (2019): 7.583 Concentration and Frequency Dependent Dielectric Permittivity and Dielectric Loss Study of Brucine - Chloroform Solution using Time Domain Reflectometry (TDR) A. R. Lathi Associate Professor, A.E.S. College, Hingoli, Maharashtra, India Abstract: Dielectric permittivity (ε’) and dielectric loss (ε’’) of Brucine-Chloroform solution over a frequency range of 10 MHz to 30 GHz have been measured using Time Domain Reflectometry (TDR). Observations are taken for different molar concentrations of Brucine-Chloroform (0M to 0.3M) at 298°K. Step pulse without sample R1(t) and with sample Rx(t) were recorded in time window of 5nS and digitized in 2000 points. The effect of concentration and frequency on dielectric permittivity and dielectric loss has been presented in this paper. Keywords: Dielectric permittivity; Dielectric Loss; Brucine; TDR 1. Introduction 2.2 Experimental Setup Brucine is a natural alkaloid which comes under Indole The time domain Reflectometry is used to describe a Group II [1]. Brucine was discovered in 1819 by Pelletier technique of observing time dependence reflection response and Caventoy in the bark of Strychnos nux-vomica tree. The of sample under study. H.Fellmer - Feldegg [7] developed structure of Brucine related to Strychnine [2]. Brucine TDR for measurement of dielectric loss and dielectric having anti-tumor effect [3]. It is also used in Chinese permittivity at very high frequency range. The schematic medicine as analgesic agent[4]. Brucine has antiprolifrative block diagram of the experimental setup of TDR [8,9] is effects in different cancer cell lines [5]. -

Cigarette Additives, Carcinogens and Chemicals Nicotine
Cigarette Additives, Carcinogens and Chemicals Nicotine A Destructive Natural Pesticide Which ... Is extremely addictive when smoked Is extremely addictive when chewed Causes addiction as permanent as Is harder to quit than heroin or cocaine alcoholism Is not medicine and its use not therapy Is ineffective as a stand-alone quitting aid Prevents pre-cancerous cells from dying Accelerates cancer tumor growth rates Contributes to artery hardening Has a metabolite which may cause cancer May kill brain cells and impair memory Is linked to lung cancer Likely causes brain damage and Is also a fetus destroying teratogen depression Kills half of adult smokers 13-14 years Is beat by never taking another puff or early chew! 81 Cancer Causing Chemicals Have So Far Been Identified in Cigarettes Acetaldehyde Acetamide Acrylamide Acrylonitrile 2-Amino-3,4-dimethyl-3H-imidazo[4,5-f]quinoline (MeIQ) 3-Amino-1,4-dimethyl-5H-pyrido [4,3-b]indole (Trp-P-1) 2-Amino-l-methyl-6-phenyl-1H-imidazo [4,5-b]pyridine (PhlP) 2-Amino-6-methyldipyrido[1,2-a:3',2'-d]imidazole (Glu-P-1) 3-Amino-l-methyl-5H-pyrido {4,3-b]indole (Trp-P-2 2-Amino-3-methyl-9H-pyrido[2,3-b]indole (MeAaC) 2-Amino-9H-pyrido[2,3-b]indole (AaC) 4-Aminobiphenyl 2-Aminodipyrido[1,2-a:3',2'-d]imidazole (Glu-P-2) 0-Anisidine Arsenic Benz[a]anthracene Benzene Benzo[a]pyrene Benzo[b]fluoranthene Benzo[j]fluoranthene Benzo[k]fluoranthene Benzo[b]furan Beryllium 1,3-Butadiene Cadmium Catechol (1,2-benzenediol) p-Chloroaniline Chloroform Cobalt p,p'-DDT Dibenz[a,h]acridine Dibenz[a,j]acridine Dibenz(a,h)anthracene -

The Anti-Tumor Effects of Alkaloids from the Seeds of Strychnos Nux-Vomica
Journal of Ethnopharmacology 106 (2006) 179–186 The anti-tumor effects of alkaloids from the seeds of Strychnos nux-vomica on HepG2 cells and its possible mechanism Xu-Kun Deng a,1,WuYinb,1, Wei-Dong Li a, Fang-Zhou Yin a, Xiao-Yu Lu a, Xiao-Chun Zhang a, Zi-Chun Hua b, Bao-Chang Cai a,∗ a College of Pharmacy, Nanjing University of Chinese Medicine, Nanjing 210029, China b State Key Lab of Pharmaceutic Biotechnology, Nanjing University, Nanjing 210093, China Received 6 April 2005; received in revised form 12 December 2005; accepted 15 December 2005 Available online 26 January 2006 Abstract To screen the anti-tumor effects of the four alkaloids: brucine, strychnine, brucine N-oxide and isostrychnine from the seed of Strychnos nux- vomica, MTT assay was used to examine the growth inhibitory effects of these alkaloids on human hepatoma cell line (HepG2). Brucine, strychnine and isostrychnine revealed significant inhibitory effects against HepG2 cell proliferation, whereas brucine N-oxide didn’t have such an effect. In addition, brucine caused HepG2 cell shrinkage, membrane blebbing, apoptotic body formation, all of which are typical characteristics of apoptotic programmed cell death. The results of flow cytometric analysis demonstrated that brucine caused dose-dependent apoptosis of HepG2 cells through cell cycle arrest at G0/G1 phase, thus preventing cells entering S or G2/M phase. Immunoblot results revealed that brucine significantly decreased the protein expression level of cyclooxygenase-2, whereas increased the expression caspase-3 as well as the caspase-3-like protease activity in HepG2 cells, suggesting the involvement of cyclooxygenase-2 and caspase-3 in the pro-apoptotic effects exerted by brucine. -

WIDESPREAD OCCURRENCE of SPECIFIC, HIGH AFFINITY BINDING SITES for AMINO ACIDS Stephen C
Vol. 80, No. 1, 1978 BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS January 13, 1978 Pages 161-168 WIDESPREAD OCCURRENCE OF SPECIFIC, HIGH AFFINITY BINDING SITES FOR AMINO ACIDS Stephen C. Bondy and Marilyn E. Harrington Departments of Neurology and Pharmacology University of Colorado Medical Center, 42DO East Ninth Avenue Denver, Colorado 80262 Received September 27, 1977 The high affinity binding of several amino acids to various membrane and protein preparations has been measured. Binding of radioactive amino acids suspected of being neurotransmitters and also of leucine and tyrosine to brain, liver and heart muscle membranes was saturable, reversible and stereospecific. Similar characteristics were found using chloroform-methanol extracted brain tissue and heat denatured albumin. Compounds thought to act as blockers of postsynaptic binding such as strychnine, bicuculline and kainic acid did not inhibit binding. Thus, specific high affinity interactions between amino acids and proteins are widespread and largely unrelated to neurotransmission. INTRODUCTION The affinity binding of several amino acids such as glutamic acid, glycine and gamma-aminobutyric acid has been reported in membrane preparations of brain (l-4). This binding has been used as a measure of postsynaptic sites specific for a particular amino acid neurotransmitter. Criteria used to establish the identity of these sites include: l) specificity of the interaction and also stereospecific selectivity in several cases; 2) saturable, reversible binding kinetics with a high affinity between membrane and ligand; 3) independence of binding on the presence of the sodium ion (5); 4) no energy dependence of the interaction, temperature should not have a major effect on binding velocity; 5) physiological relevance and appropriate inhibition by known antagonists. -

United States Patent Office
3,590,077 United States Patent Office Patented June 29, 1971 2 propane-1,3-diol and L(--)-threo-1-p-nitrophenyl-2-N,N- 3,590,077 dimethylamino-propane-1,3-diol and their acid addition SALTS OF CHRYSANTHEMIC ACID WITH 1-p- NITROPHENYL - 2-N,N - DIMETHYL-AMINO salts. The said diastereoisomers are unexpectedly excel PROPANE-1,3-DIOLS lent agents for the resolution of racemic acids since they Georges Muller, Nogent-sur-Marne, Gaston Amiard, form salts of greater stability and better crystallizability Thorigny, Andre Poittevin, Les Lilas, and Vesperto and the bases are less soluble in aqueous media than Torelli, Maisons Alfort, France, assignors to Roussel known resolving agents such as ephedrine or L(--)- UCLAF, Paris, France threo-1-p-nitrophenyl-2-amino-propane-1,3-diol. No Drawing. Continuation-in-part of application Ser. No. The disastereoisomers of the invention are particu 610,219, Jan. 19, 1967. This application July 5, 1968, O larly useful for the resolution of racemic mixtures of Ser. No. 742,485 Claims priority, application France, Jan. 26, 1966, 1,5-dioxo - 7a - lower alkyl-5,6,7,7a-tetrahydroindane-4- 47,305; July 7, 1967, 113,580 acetic acids which have been difficult to separate with The portion of the term of the patent subsequent to known resolution agents due to the very slight solubility Mar. 9, 1987, has been disclaimed differences and poor stability of diastereoisomeric salts Int, C. C07c 91/20 formed therewith, such as with L(--)-threo-1-p-nitro U.S. C. 260-501.7 4 Claims phenyl-2-amino-propane-1,3-diol.