Determination of Selenium in Selenium Enriched Yeast
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Transport of Dangerous Goods
ST/SG/AC.10/1/Rev.16 (Vol.I) Recommendations on the TRANSPORT OF DANGEROUS GOODS Model Regulations Volume I Sixteenth revised edition UNITED NATIONS New York and Geneva, 2009 NOTE The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the Secretariat of the United Nations concerning the legal status of any country, territory, city or area, or of its authorities, or concerning the delimitation of its frontiers or boundaries. ST/SG/AC.10/1/Rev.16 (Vol.I) Copyright © United Nations, 2009 All rights reserved. No part of this publication may, for sales purposes, be reproduced, stored in a retrieval system or transmitted in any form or by any means, electronic, electrostatic, magnetic tape, mechanical, photocopying or otherwise, without prior permission in writing from the United Nations. UNITED NATIONS Sales No. E.09.VIII.2 ISBN 978-92-1-139136-7 (complete set of two volumes) ISSN 1014-5753 Volumes I and II not to be sold separately FOREWORD The Recommendations on the Transport of Dangerous Goods are addressed to governments and to the international organizations concerned with safety in the transport of dangerous goods. The first version, prepared by the United Nations Economic and Social Council's Committee of Experts on the Transport of Dangerous Goods, was published in 1956 (ST/ECA/43-E/CN.2/170). In response to developments in technology and the changing needs of users, they have been regularly amended and updated at succeeding sessions of the Committee of Experts pursuant to Resolution 645 G (XXIII) of 26 April 1957 of the Economic and Social Council and subsequent resolutions. -

Assessment of Portable HAZMAT Sensors for First Responders
The author(s) shown below used Federal funds provided by the U.S. Department of Justice and prepared the following final report: Document Title: Assessment of Portable HAZMAT Sensors for First Responders Author(s): Chad Huffman, Ph.D., Lars Ericson, Ph.D. Document No.: 246708 Date Received: May 2014 Award Number: 2010-IJ-CX-K024 This report has not been published by the U.S. Department of Justice. To provide better customer service, NCJRS has made this Federally- funded grant report available electronically. Opinions or points of view expressed are those of the author(s) and do not necessarily reflect the official position or policies of the U.S. Department of Justice. Assessment of Portable HAZMAT Sensors for First Responders DOJ Office of Justice Programs National Institute of Justice Sensor, Surveillance, and Biometric Technologies (SSBT) Center of Excellence (CoE) March 1, 2012 Submitted by ManTech Advanced Systems International 1000 Technology Drive, Suite 3310 Fairmont, West Virginia 26554 Telephone: (304) 368-4120 Fax: (304) 366-8096 Dr. Chad Huffman, Senior Scientist Dr. Lars Ericson, Director UNCLASSIFIED This project was supported by Award No. 2010-IJ-CX-K024, awarded by the National Institute of Justice, Office of Justice Programs, U.S. Department of Justice. The opinions, findings, and conclusions or recommendations expressed in this publication are those of the author(s) and do not necessarily reflect those of the Department of Justice. This document is a research report submitted to the U.S. Department of Justice. This report has not been published by the Department. Opinions or points of view expressed are those of the author(s) and do not necessarily reflect the official position or policies of the U.S. -

Underactive Thyroid
Underactive Thyroid PDF generated using the open source mwlib toolkit. See http://code.pediapress.com/ for more information. PDF generated at: Thu, 21 Jun 2012 14:27:58 UTC Contents Articles Thyroid 1 Hypothyroidism 14 Nutrition 22 B vitamins 47 Vitamin E 53 Iodine 60 Selenium 75 Omega-6 fatty acid 90 Borage 94 Tyrosine 97 Phytotherapy 103 Fucus vesiculosus 107 Commiphora wightii 110 Nori 112 Desiccated thyroid extract 116 References Article Sources and Contributors 121 Image Sources, Licenses and Contributors 124 Article Licenses License 126 Thyroid 1 Thyroid thyroid Thyroid and parathyroid. Latin glandula thyroidea [1] Gray's subject #272 1269 System Endocrine system Precursor Thyroid diverticulum (an extension of endoderm into 2nd Branchial arch) [2] MeSH Thyroid+Gland [3] Dorlands/Elsevier Thyroid gland The thyroid gland or simply, the thyroid /ˈθaɪrɔɪd/, in vertebrate anatomy, is one of the largest endocrine glands. The thyroid gland is found in the neck, below the thyroid cartilage (which forms the laryngeal prominence, or "Adam's apple"). The isthmus (the bridge between the two lobes of the thyroid) is located inferior to the cricoid cartilage. The thyroid gland controls how quickly the body uses energy, makes proteins, and controls how sensitive the body is to other hormones. It participates in these processes by producing thyroid hormones, the principal ones being triiodothyronine (T ) and thyroxine which can sometimes be referred to as tetraiodothyronine (T ). These hormones 3 4 regulate the rate of metabolism and affect the growth and rate of function of many other systems in the body. T and 3 T are synthesized from both iodine and tyrosine. -

1/2 Toxic Compound Data Sheet Name: Indene CAS Number: 00095
1/2 Toxic Compound Data Sheet Name: Indene CAS Number: 00095-13-6 Justification: This compound is listed in Ohio Administrative Code 3745 - 114 - 01 because it fulfills one or more of the following criteria: substances that are known to be, or may reasonably be anticipated to be, carcinogenic, mutagenic, teratogenic, or neurotoxic, causes reproductive dysfunction, is acutely or chronically toxic, or causes the threat of adverse environmental effects through ambient concentrations, bioaccumulation, or atmospheric deposition. lndene is acutely toxic with effects on the upper respiratory system (mucous membranes), pulmonary irritation, liver and kidney effects. Molecular Weight (g/mol): 116.15 Synonyms: Indonaphthene U.S. EPA Carcinogenic Classification (IRIS): Not listed on IRIS. PBT: Not listed as persistent, bioaccumulative and toxic. NTP: Not listed by the National Toxicology Program. HAP: Not listed as a hazardous air pollutant (HAP) by U.S. EPA. 112r: Not listed under Section 112(r) of the Clean Air Act. ACGIH: TLV: 10 ppm or 47,505 µg/m3. Critical effects include upper respiratory irritation or damage, pulmonary irritation, and liver and kidney effects. HSDB: Listed in the Hazardous Substances Data Bank. Inhalation of indene vapors is expected to cause irritation of mucous membranes. International IARC: Not listed by International Agency for Research on Cancer (IARC). ATSDR (MRL): Not listed by ATSDR. DataSheet Indene.wpd 2/2 Reference Material 1. American Conference of Governmental Industrial Hygienists (ACGIH) 2006. TLVs and BEIs: -

Selenium Hexafluoride INTERIM 1: 11-2007 1 2 3 INTERIM ACUTE EXPOSURE GUIDELINE LEVELS (Aegls) 5 for 6 Selenium Hexafluoride 7 (CAS Reg
Selenium Hexafluoride INTERIM 1: 11-2007 1 2 3 INTERIM ACUTE EXPOSURE GUIDELINE LEVELS (AEGLs) 5 FOR 6 Selenium Hexafluoride 7 (CAS Reg. No. 7783-79-1) 8 9 Se-F6 10 11 12 13 14 15 16 17 18 19 20 21 22 Selenium Hexafluoride INTERIM 1: 11-2007 1 INTERIM ACUTE EXPOSURE GUIDELINE LEVELS (AEGLs) 3 FOR 4 SELENIUM HEXAFLUORIDE 5 (CAS Reg. No. 7783-79-1) 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 4 Selenium Hexafluoride INTERIM 1: 11-2007 1 2 3 4 5 PREFACE 6 7 Under the authority of the Federal Advisory Committee Act (FACA) P. L. 92-463 of 8 1972, the National Advisory Committee for Acute Exposure Guideline Levels for Hazardous 9 Substances (NAC/AEGL Committee) has been established to identify, review and interpret 10 relevant toxicologic and other scientific data and develop AEGLs for high priority, acutely toxic 11 chemicals. 12 13 AEGLs represent threshold exposure limits for the general public and are applicable to 14 emergency exposure periods ranging from 10 minutes to 8 hours. Three levels C AEGL-1, 15 AEGL-2 and AEGL-3 C are developed for each of five exposure periods (10 and 30 minutes, 1 16 hour, 4 hours, and 8 hours) and are distinguished by varying degrees of severity of toxic effects. 17 The three AEGLs are defined as follows: 18 19 AEGL-1 is the airborne concentration (expressed as parts per million or milligrams per 20 cubic meter [ppm or mg/m3]) of a substance above which it is predicted that the general 21 population, including susceptible individuals, could experience notable discomfort, irritation, or 22 certain asymptomatic, non-sensory effects. -

(12) United States Patent (10) Patent No.: US 8,524,796 B2 Kim Et Al
US008524796B2 (12) United States Patent (10) Patent No.: US 8,524,796 B2 Kim et al. (45) Date of Patent: *Sep. 3, 2013 (54) ACTIVE POLYMER COMPOSITIONS WO 20060967.91 9, 2006 WO 2007024.125 3, 2007 (75) Inventors: Young-Sam Kim, Midland, MI (US); WO 20070785682007030791 3,7/2007 2007 Leonardo C. Lopez, Midland, MI (US); WO 2007099397 9, 2007 Scott T. Matteucci, Midland, MI (US); WO 2007121458 10/2007 Steven R. Lakso, Sanford, MI (US) WO 2008101051 8, 2008 WO 2008112833 9, 2008 (73) Assignee: Dow Global Technologies LLC WO 2008.150970 12/2008 WO 2009 134824 11, 2009 (*) Notice: Subject to any disclaimer, the term of this OTHER PUBLICATIONS patent is extended or adjusted under 35 U.S.C. 154(b) by 457 days. Ciferri, Alberto, “Supramolecular Polymers'. Second Edition, 2005, pp. 157-158, CRC Press. This patent is Subject to a terminal dis Corbin et al., “Chapter 6 Hydrogen-Bonded Supramolecular Poly claimer. mers: Linear and Network Polymers and Self-Assembling Discotic Polymers'. Supramolecular Polymers, 2nd edition, CRC Press, (21) Appl. No.: 12/539,793 2005, pp. 153-185. Duan et al., “Preparation of Antimicrobial Poly (e-caprolactone) (22) Filed: Aug. 12, 2009 Electrospun Nanofibers Containing Silver-Loaded Zirconium Phos phate Nanoparticles”, Journal of Applied Polymer Sciences, 2007. (65) Prior Publication Data vol. 106, pp. 1208-1214, Wiley Periodicals, Inc. US 201O/OO41292 A1 Feb. 18, 2010 Hagewood, "Potential of Polymeric Nanofibers for Nonwovens and Medical Applications'. Fiberjournal.com, Feb. 26, 2008, 4 Pages, Related U.S. Application Data J.Hagewood, LLC and Ben Shuler, Hills, Inc. -

The Health and Environmental Impacts of Hazardous Wastes
The health and environmental impacts of hazardous wastes IMPACT PROFILES Final report Prepared for: The Department of the Environment Prepared by: Ascend Waste and Environment Pty Ltd Date::7 June 2015 Geoff Latimer Project Number: Project # 15001AG The health and environmental impacts of hazardous wastes Project # 15001AG Date: 7 June 2015 This report has been prepared for The Department of the Environment in accordance with the terms and conditions of appointment dated 6 January 2015, and is based on the assumptions and exclusions set out in our scope of work. The report must not be reproduced in whole or in part except with the prior consent of Ascend Waste and Environment Pty Ltd and subject to inclusion of an acknowledgement of the source. Whilst reasonable attempts have been made to ensure that the contents of this report are accurate and complete at the time of writing, Ascend Waste and Environment Pty Ltd cannot accept any responsibility for any use of or reliance on the contents of this report by any third party. © Ascend Waste and Environment Pty Ltd VERSION CONTROL RECORD Document File Name Date Issued Version Author Reviewer Draft first profiles for review 11 March 2015 Rev 0 Geoff Latimer Ian Rae 15001AG_AWE_Health Env 27 April 2015 Rev0 Geoff Latimer Ian Rae Impacts draft report rev 0 15001AG_AWE_Health Env 7 June 2015 Rev0 Geoff Latimer Ian Rae Impacts final report rev 0 The health and environmental impacts of hazardous wastes Contents Page 1 Introduction 5 2 Summary report: Australia’s key hazardous waste impacts and risks -

Cal/OSHA, DOT HAZMAT, EEOC, EPA, HIPAA, IATA, IMDG, TDG, MSHA, OSHA, Australia WHS, and Canada OHS Regulations and Safety Online Training
! Cal/OSHA, DOT HAZMAT, EEOC, EPA, HIPAA, IATA, IMDG, TDG, MSHA, OSHA, Australia WHS, and Canada OHS Regulations and Safety Online Training This document is provided as a training aid and may not reflect current laws and regulations. Be sure and consult with the appropriate governing agencies or publication providers listed in the "Resources" section of our website. www.ComplianceTrainingOnline.com ! ! ! ! ! Facebook LinkedIn Twitter Google Plus Website Hazardous Chemicals Handbook Second edition Phillip Carson PhD MSc AMCT CChem FRSC FIOSH Head of Science Support Services, Unilever Research Laboratory, Port Sunlight, UK Clive Mumford BSc PhD DSc CEng MIChemE Consultant Chemical Engineer Oxford Amsterdam Boston London New York Paris San Diego San Francisco Singapore Sydney Tokyo Butterworth-Heinemann An imprint of Elsevier Science Linacre House, Jordan Hill, Oxford OX2 8DP 225 Wildwood Avenue, Woburn, MA 01801-2041 First published 1994 Second edition 2002 Copyright © 1994, 2002, Phillip Carson, Clive Mumford. All rights reserved The right of Phillip Carson and Clive Mumford to be identified as the authors of this work has been asserted in accordance with the Copyright, Designs and Patents Act 1988 No part of this publication may be reproduced in any material form (including photocopying or storing in any medium by electronic means and whether or not transiently or incidentally to some other use of this publication) without the written permission of the copyright holder except in accordance with the provisions of the Copyright, Designs and Patents Act 1988 or under the terms of a licence issued by the Copyright Licensing Agency Ltd, 90 Tottenham Court Road, London, England W1T 4LP. -

Chemical Names and CAS Numbers Final
Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number C3H8O 1‐propanol C4H7BrO2 2‐bromobutyric acid 80‐58‐0 GeH3COOH 2‐germaacetic acid C4H10 2‐methylpropane 75‐28‐5 C3H8O 2‐propanol 67‐63‐0 C6H10O3 4‐acetylbutyric acid 448671 C4H7BrO2 4‐bromobutyric acid 2623‐87‐2 CH3CHO acetaldehyde CH3CONH2 acetamide C8H9NO2 acetaminophen 103‐90‐2 − C2H3O2 acetate ion − CH3COO acetate ion C2H4O2 acetic acid 64‐19‐7 CH3COOH acetic acid (CH3)2CO acetone CH3COCl acetyl chloride C2H2 acetylene 74‐86‐2 HCCH acetylene C9H8O4 acetylsalicylic acid 50‐78‐2 H2C(CH)CN acrylonitrile C3H7NO2 Ala C3H7NO2 alanine 56‐41‐7 NaAlSi3O3 albite AlSb aluminium antimonide 25152‐52‐7 AlAs aluminium arsenide 22831‐42‐1 AlBO2 aluminium borate 61279‐70‐7 AlBO aluminium boron oxide 12041‐48‐4 AlBr3 aluminium bromide 7727‐15‐3 AlBr3•6H2O aluminium bromide hexahydrate 2149397 AlCl4Cs aluminium caesium tetrachloride 17992‐03‐9 AlCl3 aluminium chloride (anhydrous) 7446‐70‐0 AlCl3•6H2O aluminium chloride hexahydrate 7784‐13‐6 AlClO aluminium chloride oxide 13596‐11‐7 AlB2 aluminium diboride 12041‐50‐8 AlF2 aluminium difluoride 13569‐23‐8 AlF2O aluminium difluoride oxide 38344‐66‐0 AlB12 aluminium dodecaboride 12041‐54‐2 Al2F6 aluminium fluoride 17949‐86‐9 AlF3 aluminium fluoride 7784‐18‐1 Al(CHO2)3 aluminium formate 7360‐53‐4 1 of 75 Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number Al(OH)3 aluminium hydroxide 21645‐51‐2 Al2I6 aluminium iodide 18898‐35‐6 AlI3 aluminium iodide 7784‐23‐8 AlBr aluminium monobromide 22359‐97‐3 AlCl aluminium monochloride -
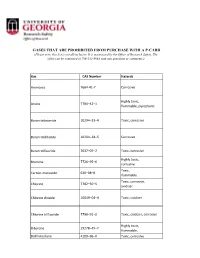
Lists of Gases for Pcard Manual.Xlsx
GASES THAT ARE PROHIBITED FROM PURCHASE WITH A P-CARD (Please note, this list is not all-inclusive. It is maintained by the Office of Research Safety. The office can be contacted at 706-542-9088 with any questions or comments.) Gas CAS Number Hazards Ammonia 7664‐41‐7 Corrosive Highly toxic, Arsine 7784–42–1 flammable, pyrophoric Boron tribromide 10294–33–4 Toxic, corrosive Boron trichloride 10294–34–5 Corrosive Boron trifluoride 7637–07–2 Toxic, corrosive Highly toxic, Bromine 7726–95–6 corrosive Toxic, Carbon monoxide 630–08–0 flammable Toxic, corrosive, Chlorine 7782–50–5 oxidizer Chlorine dioxide 10049–04–4 Toxic, oxidizer Chlorine trifluoride 7790–91–2 Toxic, oxidizer, corrosive Highly toxic, Diborane 19278–45–7 flammable, Dichlorosilane 4109–96–0 Toxic, corrosive Ethylene oxide 75–21–8 Toxic, flammable Highly toxic, corrosive, Fluorine 7782–41–4 oxidizer Highly toxic, Germane 7782–65–2 flammable Hydrogen bromide 10035–10–6 Toxic, corrosive Hydrogen chloride 7647–01–0 Toxic, corrosive Hydrogen cyanide 74–90–8 Highly toxic, flammable Hydrogen fluoride 7664–39–3 Toxic, corrosive Hydrogen iodide 10034‐85‐2 Toxic, corrosive Highly toxic, Hydrogen selenide 7783–07–5 flammable Toxic, flammable, Hydrogen sulfide 7783–06–4 corrosive Methyl bromide 74–83–9 Toxic Methyl isocyanate 624‐83‐9 Highly toxic, flammable Methyl mercaptan 74–93–1 Toxic, flammable Nickel carbonyl 13463–39–3 Highly toxic, flammable Highly toxic, Nitric oxide 10102–43–9 oxidizer Highly toxic, Nitrogen dioxide 10102–44–0 oxidizer, corrosive Highly toxic, Ozone 10028‐15‐6 -

Observations on the Rare Earths
];: H S ' . [bits sfi iiie &rc E*rlks emisiry M. S. I 9 1 5 J".J33i:tA23.V" THE UNIVERSITY OF ILLINOIS LIBRARY VMS 0F "' HE UNIVEHSITV OF UuuM OBSERVATIONS ON THE RARE EARTHS BY EDWARD WICHERS A.B. Hope College, 1913 THESIS Submitted in Partial Fulfillment of the Requirements for the Degree of MASTER OF SCIENCE IN CHEMISTRY IN THE GRADUATE SCHOOL OF THE UNIVERSITY OF ILLINOIS 1915 Digitized by the Internet Archive in 2013 http://archive.org/details/observationsonraOOwich_0 UNIVERSITY OF ILLINOIS THE GRADUATE SCHOOL 5* i9X I HEREBY RECOMMEND THAT THE THESIS PREPARED UNDER MY SUPER- VISION by gflwart Wlchera, entitled 0T> serrations on tue Bare Eartaa BE ACCEPTED AS FULFILLING THIS PART OF THE REQUIREMENTS FOR THE nxr^r.^ ™? Master of Science in cnemistry DEGREE OF _ __ _ - _ _ _ In Charge of Thesis IflC // ac 7 ^ / Head of Department Recommendation concurred in :* Committee on Final Examination* *Required for doctor's degree but not for master's. 1 OBSERVATIONS ON IEB RARE EARTHS. INTRODUCTION. The chief problem which has stood in the way of the development of the chemistry of the rare earth metals has been that of finding adequate methods for the separation of the ele- ments comprising the group. For this reason, these elements have been, up to the present time, of little other than purely scien- tific interest. However, the comparatively recent discovery of many important industrial uses for other of the rarer elements, hitherto also considered of interest only in the laboratory, in- dicates the probability of finding for the rare earth metals uses which will be no less important. -

TIH/PIH List
Hazardous Materials Designated as TIH/PIH (consolidated AAR and Railinc lists) 3/12/2007 STCC Proper Shipping Name 4921402 2-CHLOROETHANAL 4921495 2-METHYL-2-HEPTANETHIOL 4921741 3,5-DICHLORO-2,4,6-TRIFLUOROPYRIDINE 4921401 ACETONE CYANOHYDRIN, STABILIZED 4927007 ACROLEIN, STABILIZED 4921019 ALLYL ALCOHOL 4923113 ALLYL CHLOROFORMATE 4921004 ALLYLAMINE 4904211 AMMONIA SOLUTION 4920360 AMMONIA SOLUTIONS 4904209 AMMONIA, ANHYDROUS 4904210 AMMONIA, ANHYDROUS 4904879 AMMONIA, ANHYDROUS 4920359 AMMONIA, ANHYDROUS 4923209 ARSENIC TRICHLORIDE 4920135 ARSINE 4932010 BORON TRIBROMIDE 4920349 BORON TRICHLORIDE 4920522 BORON TRIFLUORIDE 4936110 BROMINE 4920715 BROMINE CHLORIDE 4918505 BROMINE PENTAFLUORIDE 4936106 BROMINE SOLUTIONS 4918507 BROMINE TRIFLUORIDE 4921727 BROMOACETONE 4920343 CARBON MONOXIDE AND HYDROGEN MIXTURE, COMPRESSED 4920399 CARBON MONOXIDE, COMPRESSED 4920511 CARBON MONOXIDE, REFRIGERATED LIQUID 4920559 CARBONYL FLUORIDE 4920351 CARBONYL SULFIDE 4920523 CHLORINE 4920189 CHLORINE PENTAFLUORIDE 4920352 CHLORINE TRIFLUORIDE 4921558 CHLOROACETONE, STABILIZED 4921009 CHLOROACETONITRILE 4923117 CHLOROACETYL CHLORIDE 4921414 CHLOROPICRIN 4920516 CHLOROPICRIN AND METHYL BROMIDE MIXTURES 4920547 CHLOROPICRIN AND METHYL BROMIDE MIXTURES 4920392 CHLOROPICRIN AND METHYL CHLORIDE MIXTURES 4921746 CHLOROPIVALOYL CHLORIDE 4930204 CHLOROSULFONIC ACID 4920527 COAL GAS, COMPRESSED 4920102 COMMPRESSED GAS, TOXIC, FLAMMABLE, CORROSIVE, N.O.S. 4920303 COMMPRESSED GAS, TOXIC, FLAMMABLE, CORROSIVE, N.O.S. 4920304 COMMPRESSED GAS, TOXIC, FLAMMABLE, CORROSIVE, N.O.S. 4920305 COMMPRESSED GAS, TOXIC, FLAMMABLE, CORROSIVE, N.O.S. 4920101 COMPRESSED GAS, TOXIC, CORROSIVE, N.O.S. 4920300 COMPRESSED GAS, TOXIC, CORROSIVE, N.O.S. 4920301 COMPRESSED GAS, TOXIC, CORROSIVE, N.O.S. 4920324 COMPRESSED GAS, TOXIC, CORROSIVE, N.O.S. 4920331 COMPRESSED GAS, TOXIC, CORROSIVE, N.O.S. 4920165 COMPRESSED GAS, TOXIC, FLAMMABLE, N.O.S. 4920378 COMPRESSED GAS, TOXIC, FLAMMABLE, N.O.S. 4920379 COMPRESSED GAS, TOXIC, FLAMMABLE, N.O.S.