Protein Structure & Folding
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Heterogeneous Impacts of Protein-Stabilizing Osmolytes On
bioRxiv preprint doi: https://doi.org/10.1101/328922; this version posted May 23, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Heterogeneous Impacts of Protein-Stabilizing Osmolytes on Hydrophobic Interaction Mrinmoy Mukherjee and Jagannath Mondal⇤ Tata Institute of Fundamental Research Hyderabad, 500107 India E-mail: [email protected],+914020203091 Abstract Osmolytes’ mechanism of protecting proteins against denaturation is a longstanding puzzle, further complicated by the complex diversities inherent in protein sequences. An emergent approach in understanding osmolytes’ mechanism of action towards biopoly- mer has been to investigate osmolytes’ interplay with hydrophobic interaction, the ma- jor driving force of protein folding. However, the crucial question is whether all these protein-stabilizing osmolytes display a single unified mechanism towards hydrophobic interactions. By simulating the hydrophobic collapse of a macromolecule in aqueous solutions of two such osmoprotectants, Glycine and Trimethyl N-oxide (TMAO), both of which are known to stabilize protein’s folded conformation, we here demonstrate that these two osmolytes can impart mutually contrasting effects towards hydropho- bic interaction. While TMAO preserves its protectant nature across diverse range of polymer-osmolyte interactions, glycine is found to display an interesting cross-over from being a protectant at weaker polymer-osmolyte interaction to a denaturant of hydrophobicity at stronger polymer-osmolyte interactions. A preferential-interaction analysis reveals that a subtle balance of conformation-dependent exclusion/binding of ⇤To whom correspondence should be addressed 1 bioRxiv preprint doi: https://doi.org/10.1101/328922; this version posted May 23, 2018. -

DNA Glycosylase Exercise - Levels 1 & 2: Answer Key
Name________________________ StarBiochem DNA Glycosylase Exercise - Levels 1 & 2: Answer Key Background In this exercise, you will explore the structure of a DNA repair protein found in most species, including bacteria. DNA repair proteins move along DNA strands, checking for mistakes or damage. DNA glycosylases, a specific type of DNA repair protein, recognize DNA bases that have been chemically altered and remove them, leaving a site in the DNA without a base. Other proteins then come along to fill in the missing DNA base. Learning objectives We will explore the relationship between a protein’s structure and its function in a human DNA glycosylase called human 8-oxoguanine glycosylase (hOGG1). Getting started We will begin this exercise by exploring the structure of hOGG1 using a molecular 3-D viewer called StarBiochem. In this particular structure, the repair protein is bound to a segment of DNA that has been damaged. We will first focus on the structure hOGG1 and then on how this protein interacts with DNA to repair a damaged DNA base. • To begin using StarBiochem, please navigate to: http://mit.edu/star/biochem/. • Click on the Start button Click on the Start button for StarBiochem. • Click Trust when a prompt appears asking if you trust the certificate. • In the top menu, click on Samples à Select from Samples. Within the Amino Acid/Proteins à Protein tab, select “DNA glycosylase hOGG1 w/ DNA – H. sapiens (1EBM)”. “1EBM” is the four character unique ID for this structure. Take a moment to look at the structure from various angles by rotating and zooming on the structure. -
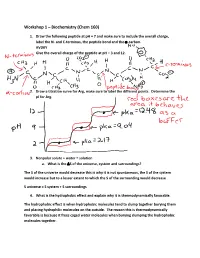
Workshop 1 – Biochemistry (Chem 160)
Workshop 1 – Biochemistry (Chem 160) 1. Draw the following peptide at pH = 7 and make sure to include the overall charge, label the N- and C-terminus, the peptide bond and the -carbon. AVDKY Give the overall charge of the peptide at pH = 3 and 12. 2. Draw a titration curve for Arg, make sure to label the different points. Determine the pI for Arg. 3. Nonpolar solute + water = solution a. What is the S of the universe, system and surroundings? The S of the universe would decrease this is why it is not spontaneous, the S of the system would increase but to a lesser extent to which the S of the surrounding would decrease S universe = S system + S surroundings 4. What is the hydrophobic effect and explain why it is thermodynamically favorable. The hydrophobic effect is when hydrophobic molecules tend to clump together burying them and placing hydrophilic molecules on the outside. The reason this is thermodynamically favorable is because it frees caged water molecules when burying clumping the hydrophobic molecules together. 5. Urea dissolves very readily in water, but the solution becomes very cold as the urea dissolves. How is this possible? Urea dissolves in water because when dissolving there is a net increase in entropy of the universe. The heat exchange, getting colder only reflects the enthalpy (H) component of the total energy change. The entropy change is high enough to offset the enthalpy component and to add up to an overall -G 6. A mutation that changes an alanine residue in the interior of a protein to valine is found to lead to a loss of activity. -

Proteasomes on the Chromosome Cell Research (2017) 27:602-603
602 Cell Research (2017) 27:602-603. © 2017 IBCB, SIBS, CAS All rights reserved 1001-0602/17 $ 32.00 RESEARCH HIGHLIGHT www.nature.com/cr Proteasomes on the chromosome Cell Research (2017) 27:602-603. doi:10.1038/cr.2017.28; published online 7 March 2017 Targeted proteolysis plays an this process, both in mediating homolog ligases, respectively, as RN components important role in the execution and association and in providing crossovers that impact crossover formation [5, 6]. regulation of many cellular events. that tether homologs and ensure their In the first of the two papers, Ahuja et Two recent papers in Science identify accurate segregation. Meiotic recom- al. [7] report that budding yeast pre9∆ novel roles for proteasome-mediated bination is initiated by DNA double- mutants, which lack a nonessential proteolysis in homologous chromo- strand breaks (DSBs) at many sites proteasome subunit, display defects in some pairing, recombination, and along chromosomes, and the multiple meiotic DSB repair, in chromosome segregation during meiosis. interhomolog interactions formed by pairing and synapsis, and in crossover Protein degradation by the 26S pro- DSB repair drive homolog association, formation. Similar defects are seen, teasome drives a variety of processes culminating in the end-to-end homolog but to a lesser extent, in cells treated central to the cell cycle, growth, and dif- synapsis by a protein structure called with the proteasome inhibitor MG132. ferentiation. Proteins are targeted to the the synaptonemal complex (SC) [3]. These defects all can be ascribed to proteasome by covalently ligated chains SC-associated focal protein complexes, a failure to remove nonhomologous of ubiquitin, a small (8.5 kDa) protein. -

Chapter 13 Protein Structure Learning Objectives
Chapter 13 Protein structure Learning objectives Upon completing this material you should be able to: ■ understand the principles of protein primary, secondary, tertiary, and quaternary structure; ■use the NCBI tool CN3D to view a protein structure; ■use the NCBI tool VAST to align two structures; ■explain the role of PDB including its purpose, contents, and tools; ■explain the role of structure annotation databases such as SCOP and CATH; and ■describe approaches to modeling the three-dimensional structure of proteins. Outline Overview of protein structure Principles of protein structure Protein Data Bank Protein structure prediction Intrinsically disordered proteins Protein structure and disease Overview: protein structure The three-dimensional structure of a protein determines its capacity to function. Christian Anfinsen and others denatured ribonuclease, observed rapid refolding, and demonstrated that the primary amino acid sequence determines its three-dimensional structure. We can study protein structure to understand problems such as the consequence of disease-causing mutations; the properties of ligand-binding sites; and the functions of homologs. Outline Overview of protein structure Principles of protein structure Protein Data Bank Protein structure prediction Intrinsically disordered proteins Protein structure and disease Protein primary and secondary structure Results from three secondary structure programs are shown, with their consensus. h: alpha helix; c: random coil; e: extended strand Protein tertiary and quaternary structure Quarternary structure: the four subunits of hemoglobin are shown (with an α 2β2 composition and one beta globin chain high- lighted) as well as four noncovalently attached heme groups. The peptide bond; phi and psi angles The peptide bond; phi and psi angles in DeepView Protein secondary structure Protein secondary structure is determined by the amino acid side chains. -

Hydrophobic Effect, Water Structure, and Heat Capacity Changes
J. Phys. Chem. B 1997, 101, 4343-4348 4343 Hydrophobic Effect, Water Structure, and Heat Capacity Changes Kim A. Sharp* and Bhupinder Madan Department of Biochemistry & Biophysics, UniVersity of PennsylVania, 3700 Hamilton Walk, Philadelphia, PennsylVania 19104-6059 ReceiVed: January 16, 1997; In Final Form: April 1, 1997X hyd The hydration heat capacity (∆Cp ) of nine solutes of varying hydrophobicity was studied using a combination of a random network model equation of water and Monte Carlo simulations. Nonpolar solutes cause a concerted decrease in the average length and angle of the water-water hydrogen bonds in the first hydration shell, while polar and ionic solutes have the opposite effect. This is due to changes in the amounts, relative to bulk water, of two populations of hydrogen bonds: one with shorter and more linear bonds and the other with longer and more bent bonds. Heat capacity changes were calculated from these changes in water structure using a random network model equation of state. The calculated changes account for observed hyd differences in ∆Cp for the various solutes. The simulations provide a unified picture of hydrophobic and hyd polar hydration, and both a structural and thermodynamic explanation of the opposite signs of ∆Cp observed for polar and nonpolar solutes. Introduction nonpolar groups means that at higher temperatures (T > 80 °C) their insolubility results from unfavorable changes in enthalpy, In recent years analysis of heat capacity changes has become not entropy. Second, the hydration of a majority of polar and of central importance in understanding the thermodynamics of ionic solutes is also accompanied by a decrease in entropy, and protein folding, protein-protein binding, protein-nucleic acid thus by some kind of structuring of water, but in this case binding, and the hydrophobic effect.1-7 There are several hyd 7,16-20 reasons for this. -

Introduction to Proteins and Amino Acids Introduction
Introduction to Proteins and Amino Acids Introduction • Twenty percent of the human body is made up of proteins. Proteins are the large, complex molecules that are critical for normal functioning of cells. • They are essential for the structure, function, and regulation of the body’s tissues and organs. • Proteins are made up of smaller units called amino acids, which are building blocks of proteins. They are attached to one another by peptide bonds forming a long chain of proteins. Amino acid structure and its classification • An amino acid contains both a carboxylic group and an amino group. Amino acids that have an amino group bonded directly to the alpha-carbon are referred to as alpha amino acids. • Every alpha amino acid has a carbon atom, called an alpha carbon, Cα ; bonded to a carboxylic acid, –COOH group; an amino, –NH2 group; a hydrogen atom; and an R group that is unique for every amino acid. Classification of amino acids • There are 20 amino acids. Based on the nature of their ‘R’ group, they are classified based on their polarity as: Classification based on essentiality: Essential amino acids are the amino acids which you need through your diet because your body cannot make them. Whereas non essential amino acids are the amino acids which are not an essential part of your diet because they can be synthesized by your body. Essential Non essential Histidine Alanine Isoleucine Arginine Leucine Aspargine Methionine Aspartate Phenyl alanine Cystine Threonine Glutamic acid Tryptophan Glycine Valine Ornithine Proline Serine Tyrosine Peptide bonds • Amino acids are linked together by ‘amide groups’ called peptide bonds. -

Structure of the Lifeact–F-Actin Complex
bioRxiv preprint doi: https://doi.org/10.1101/2020.02.16.951269; this version posted February 16, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Structure of the Lifeact–F-actin complex 2 Alexander Belyy1, Felipe Merino1,2, Oleg Sitsel1 and Stefan Raunser1* 3 1Department of Structural Biochemistry, Max Planck Institute of Molecular Physiology, Otto-Hahn-Str. 11, 44227 Dortmund, 4 Germany 5 2Current address: Department of Protein Evolution, Max Planck Institute for Developmental Biology, Max-Planck-Ring 5, 6 72076, Tübingen, Germany. 7 *Correspondence should be addressed to: [email protected] 8 9 Abstract 10 Lifeact is a short actin-binding peptide that is used to visualize filamentous actin (F-actin) 11 structures in live eukaryotic cells using fluorescence microscopy. However, this popular probe 12 has been shown to alter cellular morphology by affecting the structure of the cytoskeleton. The 13 molecular basis for such artefacts is poorly understood. Here, we determined the high- 14 resolution structure of the Lifeact–F-actin complex using electron cryo-microscopy. The 15 structure reveals that Lifeact interacts with a hydrophobic binding pocket on F-actin and 16 stretches over two adjacent actin subunits, stabilizing the DNase I-binding loop of actin in the 17 closed conformation. Interestingly, the hydrophobic binding site is also used by actin-binding 18 proteins, such as cofilin and myosin and actin-binding toxins, such as TccC3HVR from 19 Photorhabdus luminescens and ExoY from Pseudomonas aeruginosa. -

Overcharging of Zinc Ion in the Structure of Zinc−Finger Protein Is
Overcharging of zinc ion in the structure of zinc−finger protein is needed for DNA binding stability Ly H. Nguyen,z Tuyen T. Tran,z Truong Thi Ngoc Lien,x Mai Hong Hanh,z and Toan T. Nguyen∗,z zKey Laboratory for Multiscale Simulations of Complex Systems, VNU University of Science, Vietnam National University, Hanoi, 334 Nguyen Trai street, Thanh Xuan district, Hanoi, Vietnam xHanoi University of Science and Technology, 1 Dai Co Viet street, Bach Khoa, Hai Ba Trung district, Hanoi, Vietnam E-mail: [email protected],[email protected] Running header Zinc−finger overcharging arXiv:1911.10452v2 [q-bio.BM] 22 Feb 2020 1 Abstract Zinc finger structure, where a Zn2+ ion binds to four 4 cysteines or histidines in a tetrahedral structure, is a very common motif of nucleic acid−binding proteins. The corresponding interaction model is present in 3% of the genes in human genome. As a result, zinc−finger has been extremely useful in various therapeutic and research capacities, and in biotechnology. In stable configuration of zinc−finger, the cysteine amino acids are deprotonated and become negatively charged. Thus, the Zn2+ ion is overscreened by four cysteine charges (overcharged). Whether this overcharged config- uration is also stable when such a negatively charged zinc−finger binds to a negatively charged DNA molecule is unknown. We investigated how the deprotonated state of cysteine influences its structure, dynamics, and function in binding to DNA molecules by using an all−atom molecular dynamics simulation up to microsecond range of an androgen receptor protein dimer. Our results showed that the deprotonated state of cysteine residues is essential for mechanical stabilization of the functional, folded con- formation. -

The Structure and Function of Large Biological Molecules 5
The Structure and Function of Large Biological Molecules 5 Figure 5.1 Why is the structure of a protein important for its function? KEY CONCEPTS The Molecules of Life Given the rich complexity of life on Earth, it might surprise you that the most 5.1 Macromolecules are polymers, built from monomers important large molecules found in all living things—from bacteria to elephants— can be sorted into just four main classes: carbohydrates, lipids, proteins, and nucleic 5.2 Carbohydrates serve as fuel acids. On the molecular scale, members of three of these classes—carbohydrates, and building material proteins, and nucleic acids—are huge and are therefore called macromolecules. 5.3 Lipids are a diverse group of For example, a protein may consist of thousands of atoms that form a molecular hydrophobic molecules colossus with a mass well over 100,000 daltons. Considering the size and complexity 5.4 Proteins include a diversity of of macromolecules, it is noteworthy that biochemists have determined the detailed structures, resulting in a wide structure of so many of them. The image in Figure 5.1 is a molecular model of a range of functions protein called alcohol dehydrogenase, which breaks down alcohol in the body. 5.5 Nucleic acids store, transmit, The architecture of a large biological molecule plays an essential role in its and help express hereditary function. Like water and simple organic molecules, large biological molecules information exhibit unique emergent properties arising from the orderly arrangement of their 5.6 Genomics and proteomics have atoms. In this chapter, we’ll first consider how macromolecules are built. -

Biological Membranes
14 Biological Membranes To understand the structure The fundamental unit of life is the cell. All living things are composed of Goal and composition of biological cells, be it a single cell in the case of many microorganisms or a highly membranes. organized ensemble of myriad cell types in the case of multicellular organisms. A defining feature of the cell is a membrane, the cytoplasmic Objectives membrane, that surrounds the cell and separates the inside of the cell, the After this chapter, you should be able to cytoplasm, from other cells and the extracellular milieu. Membranes also • distinguish between cis and trans surround specialized compartments inside of cells known as organelles. unsaturated fatty acids. Whereas cells are typically several microns (μm) in diameter (although • explain why phospholipids some cells can be much larger), the membrane is only about 10 nanometers spontaneously form lipid bilayers and (nm) thick. Yet, and as we will see in subsequent chapters, the membrane is sealed compartments. not simply an ultra-thin, pliable sheet that encases the cytoplasm. Rather, • describe membrane fluidity and how it membranes are dynamic structures that mediate many functions in the is affected by membrane composition life of the cell. In this chapter we examine the composition of membranes, and temperature. their assembly, the forces that stabilize them, and the chemical and physical • explain the role of cholesterol in properties that influence their function. buffering membrane fluidity. The preceding chapters have focused on two kinds of biological molecules, • explain how the polar backbone namely proteins and nucleic acids, that are important in the workings of a membrane protein can be accommodated in a bilayer. -

Hydrophobic Catalysis and a Potential Biological Role of DNA Unstacking Induced by Environment Effects
Hydrophobic catalysis and a potential biological role of DNA unstacking induced by environment effects Bobo Fenga,1, Robert P. Sosab,c,d, Anna K. F. Mårtenssona, Kai Jiange, Alex Tongf, Kevin D. Dorfmang, Masayuki Takahashih, Per Lincolna, Carlos J. Bustamanteb,c,d,f,i,j,k,l, Fredrik Westerlunde, and Bengt Nordéna,1 aDepartment of Chemistry and Chemical Engineering, Chalmers University of Technology, 412 96 Gothenburg, Sweden; bBiophysics Graduate Group, University of California, Berkeley, CA 94720; cJason L. Choy Laboratory of Single Molecule Biophysics, University of California, Berkeley, CA 94720; dInstitute for Quantitative Biosciences-QB3, University of California, Berkeley, CA 94720; eDepartment of Biology and Biological Engineering, Chalmers University of Technology, 412 96 Gothenburg, Sweden; fDepartment of Chemistry, University of California, Berkeley, CA 94720; gDepartment of Chemical Engineering and Materials Science, University of Minnesota, Twin Cities, Minneapolis, MN 55455; hSchool of Life Science and Technology, Tokyo Institute of Technology, Tokyo 152-8550, Japan; iDepartment of Molecular & Cell Biology, University of California, Berkeley, CA 94720; jDepartment of Physics, University of California, Berkeley, CA 94720; kHoward Hughes Medical Institute, University of California, Berkeley, CA 94720; and lKavli Energy Nanosciences Institute, University of California, Berkeley, CA 94720 Edited by Feng Zhang, Broad Institute, Cambridge, MA, and approved July 22, 2019 (received for review May 27, 2019) Hydrophobic base stacking