Probing the Base Stacking Contributions During
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Heterogeneous Impacts of Protein-Stabilizing Osmolytes On
bioRxiv preprint doi: https://doi.org/10.1101/328922; this version posted May 23, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Heterogeneous Impacts of Protein-Stabilizing Osmolytes on Hydrophobic Interaction Mrinmoy Mukherjee and Jagannath Mondal⇤ Tata Institute of Fundamental Research Hyderabad, 500107 India E-mail: [email protected],+914020203091 Abstract Osmolytes’ mechanism of protecting proteins against denaturation is a longstanding puzzle, further complicated by the complex diversities inherent in protein sequences. An emergent approach in understanding osmolytes’ mechanism of action towards biopoly- mer has been to investigate osmolytes’ interplay with hydrophobic interaction, the ma- jor driving force of protein folding. However, the crucial question is whether all these protein-stabilizing osmolytes display a single unified mechanism towards hydrophobic interactions. By simulating the hydrophobic collapse of a macromolecule in aqueous solutions of two such osmoprotectants, Glycine and Trimethyl N-oxide (TMAO), both of which are known to stabilize protein’s folded conformation, we here demonstrate that these two osmolytes can impart mutually contrasting effects towards hydropho- bic interaction. While TMAO preserves its protectant nature across diverse range of polymer-osmolyte interactions, glycine is found to display an interesting cross-over from being a protectant at weaker polymer-osmolyte interaction to a denaturant of hydrophobicity at stronger polymer-osmolyte interactions. A preferential-interaction analysis reveals that a subtle balance of conformation-dependent exclusion/binding of ⇤To whom correspondence should be addressed 1 bioRxiv preprint doi: https://doi.org/10.1101/328922; this version posted May 23, 2018. -

Protein Structure & Folding
6 Protein Structure & Folding To understand protein folding In the last chapter we learned that proteins are composed of amino acids Goal as a chemical equilibrium. linked together by peptide bonds. We also learned that the twenty amino acids display a wide range of chemical properties. In this chapter we will see Objectives that how a protein folds is determined by its amino acid sequence and that the three-dimensional shape of a folded protein determines its function by After this chapter, you should be able to: the way it positions these amino acids. Finally, we will see that proteins fold • describe the four levels of protein because doing so minimizes Gibbs free energy and that this minimization structure and the thermodynamic involves both making the most favorable bonds and maximizing disorder. forces that stabilize them. • explain how entropy (S) and enthalpy Proteins exhibit four levels of structure (H) contribute to Gibbs free energy. • use the equation ΔG = ΔH – TΔS The structure of proteins can be broken down into four levels of to determine the dependence of organization. The first is primary structure, the linear sequence of amino the favorability of a reaction on acids in the polypeptide chain. By convention, the primary sequence is temperature. written in order from the amino acid at the N-terminus (by convention • explain the hydrophobic effect and its usually on the left) to the amino acid at the C-terminus. The second level role in protein folding. of protein structure, secondary structure, is the local conformation adopted by stretches of contiguous amino acids. -
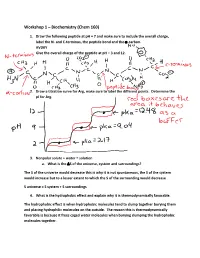
Workshop 1 – Biochemistry (Chem 160)
Workshop 1 – Biochemistry (Chem 160) 1. Draw the following peptide at pH = 7 and make sure to include the overall charge, label the N- and C-terminus, the peptide bond and the -carbon. AVDKY Give the overall charge of the peptide at pH = 3 and 12. 2. Draw a titration curve for Arg, make sure to label the different points. Determine the pI for Arg. 3. Nonpolar solute + water = solution a. What is the S of the universe, system and surroundings? The S of the universe would decrease this is why it is not spontaneous, the S of the system would increase but to a lesser extent to which the S of the surrounding would decrease S universe = S system + S surroundings 4. What is the hydrophobic effect and explain why it is thermodynamically favorable. The hydrophobic effect is when hydrophobic molecules tend to clump together burying them and placing hydrophilic molecules on the outside. The reason this is thermodynamically favorable is because it frees caged water molecules when burying clumping the hydrophobic molecules together. 5. Urea dissolves very readily in water, but the solution becomes very cold as the urea dissolves. How is this possible? Urea dissolves in water because when dissolving there is a net increase in entropy of the universe. The heat exchange, getting colder only reflects the enthalpy (H) component of the total energy change. The entropy change is high enough to offset the enthalpy component and to add up to an overall -G 6. A mutation that changes an alanine residue in the interior of a protein to valine is found to lead to a loss of activity. -

Cancer Drug Pharmacology Table
CANCER DRUG PHARMACOLOGY TABLE Cytotoxic Chemotherapy Drugs are classified according to the BC Cancer Drug Manual Monographs, unless otherwise specified (see asterisks). Subclassifications are in brackets where applicable. Alkylating Agents have reactive groups (usually alkyl) that attach to Antimetabolites are structural analogues of naturally occurring molecules DNA or RNA, leading to interruption in synthesis of DNA, RNA, or required for DNA and RNA synthesis. When substituted for the natural body proteins. substances, they disrupt DNA and RNA synthesis. bendamustine (nitrogen mustard) azacitidine (pyrimidine analogue) busulfan (alkyl sulfonate) capecitabine (pyrimidine analogue) carboplatin (platinum) cladribine (adenosine analogue) carmustine (nitrosurea) cytarabine (pyrimidine analogue) chlorambucil (nitrogen mustard) fludarabine (purine analogue) cisplatin (platinum) fluorouracil (pyrimidine analogue) cyclophosphamide (nitrogen mustard) gemcitabine (pyrimidine analogue) dacarbazine (triazine) mercaptopurine (purine analogue) estramustine (nitrogen mustard with 17-beta-estradiol) methotrexate (folate analogue) hydroxyurea pralatrexate (folate analogue) ifosfamide (nitrogen mustard) pemetrexed (folate analogue) lomustine (nitrosurea) pentostatin (purine analogue) mechlorethamine (nitrogen mustard) raltitrexed (folate analogue) melphalan (nitrogen mustard) thioguanine (purine analogue) oxaliplatin (platinum) trifluridine-tipiracil (pyrimidine analogue/thymidine phosphorylase procarbazine (triazine) inhibitor) -

The Nutrition and Food Web Archive Medical Terminology Book
The Nutrition and Food Web Archive Medical Terminology Book www.nafwa. -

The Biochemistry of Gout: a USMLE Step 1 Study Aid
The Biochemistry of Gout: A USMLE Step 1 Study Aid BMS 6204 May 26, 2005 Compiled by: Todd Kerensky Elizabeth Ballard Brendan Prendergast Eric Ritchie 1 Introduction Gout is a systemic disease caused by excess uric acid as the result of deficient purine metabolism. Clinically, gout is marked by peripheral arthritis and painful inflammation in joints resulting from deposition of uric acid in joint synovia as monosodium urate crystals. Although gout is the most common crystal-induced arthritis, a condition known as pseudogout can commonly be mistaken for gout in the clinic. Pseudogout results from deposition of calcium pyrophosphatase (CPP) crystals in synovial spaces, but causes nearly identical clinical presentation. Clinical findings Crystal-induced arthritis such as gout and pseudogout differ from other types of arthritis in their clinical presentations. The primary feature differentiating gout from other types of arthritis is the spontaneity and abruptness of onset of inflammation. Additionally, the inflammation from gout and pseudogout are commonly found in a single joint. Gout and pseudogout typically present with Podagra, a painful inflammation of the metatarsal- phalangeal joint of the great toe. However, gout can also present with spontaneous edema and painful inflammation of any other joint, but most commonly the ankle, wrist, or knee. As an exception, a spontaneous painful inflammation in the glenohumeral joint is usually the result of pseudogout. It is important to recognize the clinical differences between gout, pseudogout and other types of arthritis because the treatments differ markedly (Kaplan 2005). Pathophysiology and Treatment of Gout Although gout affects peripheral joints in clinical presentation, it is important to recognize that it is a systemic disorder caused by either overproduction or underexcretion of uric acid. -

Analysis of Tautomeric Equilibrium of the Analogue D(Dinitro-Tc(O))TP During Incorporation by the Klenow Fragment of DNA Polymerase I
Analysis of Tautomeric Equilibrium of the Analogue d(dinitro-tC(O))TP During Incorporation by the Klenow Fragment of DNA Polymerase I Arielle Baker Monday, April 7th, 2014: 4:00pm Thesis Advisor Robert Kuchta, Ph.D. | Dept. of Chemistry and Biochemistry Committee Members Robert Knight, Ph.D. | Dept. of Chemistry and Biochemistry Jennifer Martin, Ph.D. | Dept. of Molecular, Cellular and Developmental Biology This thesis is submitted in partial fulfillment of the requirements for graduation with honors in the Department of Molecular, Cellular and Developmental Biology at the University of Colorado, Boulder. Acknowledgements My most heartfelt thanks goes to my advisor, mentor and friend Dr. Rob Kuchta, for his unending patience and willingness to share his knowledge with me, as well as his continued support for my pursuit of science. I would like to thank my friends in the Kuchta lab, and to some colleagues in particular. Thank you to Dr. Andrew Olsen and Dr. Gudrun Stengel for being the very first overseers of my work, and for not scaring me off. Thanks to Dr. Ashwani Vashishtha, Dr. Joshua Gosling, Sarah Dickerson, Clarinda Hougen and Taylor Minckley for the many laughs we’ve shared benchside, and for answering even the most insignificant of questions. Thank you to my committee members, Dr. Jennifer Martin and Dr. Rob Knight, for their assistance in navigating my thesis work. My work would not have been possible without generous funding from the National Institutes of Health. Thank you to my dear friend Makenna Morck for always being available to discuss biochemistry with me in the wee hours of the morning. -

Hydrophobic Effect, Water Structure, and Heat Capacity Changes
J. Phys. Chem. B 1997, 101, 4343-4348 4343 Hydrophobic Effect, Water Structure, and Heat Capacity Changes Kim A. Sharp* and Bhupinder Madan Department of Biochemistry & Biophysics, UniVersity of PennsylVania, 3700 Hamilton Walk, Philadelphia, PennsylVania 19104-6059 ReceiVed: January 16, 1997; In Final Form: April 1, 1997X hyd The hydration heat capacity (∆Cp ) of nine solutes of varying hydrophobicity was studied using a combination of a random network model equation of water and Monte Carlo simulations. Nonpolar solutes cause a concerted decrease in the average length and angle of the water-water hydrogen bonds in the first hydration shell, while polar and ionic solutes have the opposite effect. This is due to changes in the amounts, relative to bulk water, of two populations of hydrogen bonds: one with shorter and more linear bonds and the other with longer and more bent bonds. Heat capacity changes were calculated from these changes in water structure using a random network model equation of state. The calculated changes account for observed hyd differences in ∆Cp for the various solutes. The simulations provide a unified picture of hydrophobic and hyd polar hydration, and both a structural and thermodynamic explanation of the opposite signs of ∆Cp observed for polar and nonpolar solutes. Introduction nonpolar groups means that at higher temperatures (T > 80 °C) their insolubility results from unfavorable changes in enthalpy, In recent years analysis of heat capacity changes has become not entropy. Second, the hydration of a majority of polar and of central importance in understanding the thermodynamics of ionic solutes is also accompanied by a decrease in entropy, and protein folding, protein-protein binding, protein-nucleic acid thus by some kind of structuring of water, but in this case binding, and the hydrophobic effect.1-7 There are several hyd 7,16-20 reasons for this. -

Purine Analogue 6-Methylmercaptopurine Riboside Inhibits Early and Late Phases of the Angiogenesis Process1
[CANCER RESEARCH 59, 2417–2424, May 15, 1999] Purine Analogue 6-Methylmercaptopurine Riboside Inhibits Early and Late Phases of the Angiogenesis Process1 Marco Presta,2 Marco Rusnati, Mirella Belleri, Lucia Morbidelli, Marina Ziche, and Domenico Ribatti Unit of General Pathology and Immunology, Department of Biomedical Sciences and Biotechnology, School of Medicine, University of Brescia, 25123 Brescia [M. P., M. R., M. B.]; Centro Interuniversitario di Medicina Molecolare e Biofisica Applicata (C.I.M.M.B.A.), Department of Pharmacology, University of Florence, 50134 Florence [L. M., M. Z.]; and Institute of Human Anatomy, Histology, and General Embryology, University of Bari, 70124 Bari [D. R.], Italy ABSTRACT purine synthesis and purine interconversion reactions, and their me- tabolites can be incorporated into nucleic acids (3). 6-TG3 and Angiogenesis has been identified as an important target for antineo- 6-MMPR also alter membrane glycoprotein synthesis (4). Purine plastic therapy. The use of purine analogue antimetabolites in combina- analogues can act as protein kinase inhibitors; 6-MMPR is a highly tion chemotherapy of solid tumors has been proposed. To assess the possibility that selected purine analogues may affect tumor neovascular- effective inhibitor of nerve growth factor-activated protein kinase N ization, 6-methylmercaptopurine riboside (6-MMPR), 6-methylmercapto- (5), and 2-AP inhibits proto-oncogene and IFN gene transcription (6). purine, 2-aminopurine, and adenosine were evaluated for the capacity to Moreover, purine analogues have found application in immunosup- inhibit angiogenesis in vitro and in vivo. 6-MMPR inhibited fibroblast pressive and antiviral therapy (3). At present, 6-MMP and 6-TG growth factor-2 (FGF2)-induced proliferation and delayed the repair of continue to be used mainly in the management of acute leukemia. -

A Comparative Study of Purinoceptors in Some Invertebrates and Vertebrates
A COMPARATIVE STUDY OF PURINOCEPTORS IN SOME INVERTEBRATES AND VERTEBRATES by Gillian Elizabeth Knight G.l.Biol Department Of Anatomy and Centre for Neuroscience, University College London, Gower Street, London WCIE 6BT Thesis submitted for the degree of Doctor of Philosophy University of London ProQuest Number: 10017490 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. uest. ProQuest 10017490 Published by ProQuest LLC(2016). Copyright of the Dissertation is held by the Author. All rights reserved. This work is protected against unauthorized copying under Title 17, United States Code. Microform Edition © ProQuest LLC. ProQuest LLC 789 East Eisenhower Parkway P.O. Box 1346 Ann Arbor, Ml 48106-1346 ABSTRACT The main objective of this thesis has been to identify and characterise responses to purine compounds in a variety of invertebrates and lower vertebrates, and to examine whether they act on receptors that resemble the purinoceptor subtypes established in mammals. Tissues were chosen largely from the gastrointestinal or cardiovascular systems. In mammalian systems adenosine and adenosine 5 ’-triphosphate (ATP) activate separate classes of purinoceptors; this also appears to be true for tissues in most invertebrate groups, but there are exceptions, for instance the hearts of the snail Helix aspeTsa, and slug Avion atev, and the snail rectum all possess purinoceptors that do not appear to distinguish the purine nucleosides from nucleotides. -

Biological Membranes
14 Biological Membranes To understand the structure The fundamental unit of life is the cell. All living things are composed of Goal and composition of biological cells, be it a single cell in the case of many microorganisms or a highly membranes. organized ensemble of myriad cell types in the case of multicellular organisms. A defining feature of the cell is a membrane, the cytoplasmic Objectives membrane, that surrounds the cell and separates the inside of the cell, the After this chapter, you should be able to cytoplasm, from other cells and the extracellular milieu. Membranes also • distinguish between cis and trans surround specialized compartments inside of cells known as organelles. unsaturated fatty acids. Whereas cells are typically several microns (μm) in diameter (although • explain why phospholipids some cells can be much larger), the membrane is only about 10 nanometers spontaneously form lipid bilayers and (nm) thick. Yet, and as we will see in subsequent chapters, the membrane is sealed compartments. not simply an ultra-thin, pliable sheet that encases the cytoplasm. Rather, • describe membrane fluidity and how it membranes are dynamic structures that mediate many functions in the is affected by membrane composition life of the cell. In this chapter we examine the composition of membranes, and temperature. their assembly, the forces that stabilize them, and the chemical and physical • explain the role of cholesterol in properties that influence their function. buffering membrane fluidity. The preceding chapters have focused on two kinds of biological molecules, • explain how the polar backbone namely proteins and nucleic acids, that are important in the workings of a membrane protein can be accommodated in a bilayer. -

Variations in Bacterial Adenosine Triphosphate Values Due to Genus and Environmental Conditions
VARIATIONS IN BACTERIAL ADENOSINE TRIPHOSPHATE VALUES DUE TO GENUS AND ENVIRONMENTAL CONDITIONS by Ruann Knox Hampson Thesis submitted to the Faculty of the Virginia Polytechnic Institute and State University in partial fulfillment of the requirements for the degree of Master of Science in Food Science and Technology APPROVED: M. D. Pierson, Chairman J. K. Palmer A. A. Yousten December, 1986 Blacksburg, Virginia VARIATIONS IN BACTERIAL ADENOSINE TRIPOSPHATE VALUES DUE TO GENUS AND ENVIRONMENTAL CONDITIONS by Ruann Knox Hampson Committee Chairman: Merle D. Pierson Food Science and Technology (ABSTRACT) Variations in ATP content in three ground beef spoilage bacteria, Lactobacillus brevis, Lactobacillus jensenii, and Pseudomonas sp. were investigated using the bioluminescence (luciferin-luciferase) assay. Environmental factors (temperature, atmosphere, pH, aeration, and phase of growth), as well as differences among genera and species, were studied in relation to their effect on cellular ATP. Variations for each of the environmental factors and bacteria were shown statistically to be significantly different at the 0.05 level. The mean ATP/cell for each of the bacteria was 2.71 fg/cell (1..:_ brevis), 2.20 fg/cell (1..:_ jensenii), and 1.36 fg/cell (Pseudomonas sp.). For all three bacteria, ATP/cell was lower and more stable throughout the culture's growth cycle at 3°c or in N2 . In general, ATP/cell increases from a lowest value in lag phase to a highest value in stationary phase. The effect of sonication on ATP/cell was tested for each bacterium at one set of factors. Sonication studies showed that L. brevis cells were clumping, especially in aged cultures.