Whole Genome Sequencing-Based Characterization of Human Genome Variation and Mutation Burden in Botswana
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

University of California, San Diego
UNIVERSITY OF CALIFORNIA, SAN DIEGO The post-terminal differentiation fate of RNAs revealed by next-generation sequencing A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Biomedical Sciences by Gloria Kuo Lefkowitz Committee in Charge: Professor Benjamin D. Yu, Chair Professor Richard Gallo Professor Bruce A. Hamilton Professor Miles F. Wilkinson Professor Eugene Yeo 2012 Copyright Gloria Kuo Lefkowitz, 2012 All rights reserved. The Dissertation of Gloria Kuo Lefkowitz is approved, and it is acceptable in quality and form for publication on microfilm and electronically: __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ Chair University of California, San Diego 2012 iii DEDICATION Ma and Ba, for your early indulgence and support. Matt and James, for choosing more practical callings. Roy, my love, for patiently sharing the ups and downs of this journey. iv EPIGRAPH It is foolish to tear one's hair in grief, as though sorrow would be made less by baldness. ~Cicero v TABLE OF CONTENTS Signature Page .............................................................................................................. iii Dedication .................................................................................................................... -

ABCC12 Monoclonal Antibody, Clone M9II-3
ABCC12 monoclonal antibody, Gene Symbol: ABCC12 clone M9II-3 Gene Alias: MGC27071, MRP9 Catalog Number: MAB6675 Gene Summary: This gene is a member of the superfamily of ATP-binding cassette (ABC) transporters Regulatory Status: For research use only (RUO) and the encoded protein contains two ATP-binding domains and 12 transmembrane regions. ABC proteins Product Description: Rat monoclonal antibody raised transport various molecules across extra- and against partial recombinant ABCC12. intracellular membranes. ABC genes are divided into Clone Name: M9II-3 seven distinct subfamilies: ABC1, MDR/TAP, MRP, ALD, OABP, GCN20, and White. This gene is a member of Immunogen: Recombinant protein corresponding to the MRP subfamily which is involved in multi-drug amino acids 690-734 of human ABCC12. resistance. This gene and another subfamily member are arranged head-to-tail on chromosome 16q12.1. Host: Rat Increased expression of this gene is associated with breast cancer. [provided by RefSeq] Reactivity: Human References: Applications: ICC, IHC-Fr, WB 1. Multidrug resistance-associated protein 9 (ABCC12) is (See our web site product page for detailed applications present in mouse and boar sperm. Ono N, Van der information) Heijden I, Scheffer GL, Van de Wetering K, Van Deemter E, De Haas M, Boerke A, Gadella BM, De Rooij Protocols: See our web site at DG, Neefjes JJ, Groothuis TA, Oomen L, Brocks L, http://www.abnova.com/support/protocols.asp or product Ishikawa T, Borst P. Ono N, Van der Heijden I, Scheffer page for detailed protocols GL, Van de Wetering K, Van Deemter E, De Haas M, Boerke A, Gadella BM, De Rooij DG, Neefjes JJ, Specificity: M9II-3 reacts with an internal epitope of Groothuis TA, Oomen L, Brocks L, Ishikawa T, Borst P. -

DNA Methylation Changes in Down Syndrome Derived Neural Ipscs Uncover Co-Dysregulation of ZNF and HOX3 Families of Transcription
Laan et al. Clinical Epigenetics (2020) 12:9 https://doi.org/10.1186/s13148-019-0803-1 RESEARCH Open Access DNA methylation changes in Down syndrome derived neural iPSCs uncover co- dysregulation of ZNF and HOX3 families of transcription factors Loora Laan1†, Joakim Klar1†, Maria Sobol1, Jan Hoeber1, Mansoureh Shahsavani2, Malin Kele2, Ambrin Fatima1, Muhammad Zakaria1, Göran Annerén1, Anna Falk2, Jens Schuster1 and Niklas Dahl1* Abstract Background: Down syndrome (DS) is characterized by neurodevelopmental abnormalities caused by partial or complete trisomy of human chromosome 21 (T21). Analysis of Down syndrome brain specimens has shown global epigenetic and transcriptional changes but their interplay during early neurogenesis remains largely unknown. We differentiated induced pluripotent stem cells (iPSCs) established from two DS patients with complete T21 and matched euploid donors into two distinct neural stages corresponding to early- and mid-gestational ages. Results: Using the Illumina Infinium 450K array, we assessed the DNA methylation pattern of known CpG regions and promoters across the genome in trisomic neural iPSC derivatives, and we identified a total of 500 stably and differentially methylated CpGs that were annotated to CpG islands of 151 genes. The genes were enriched within the DNA binding category, uncovering 37 factors of importance for transcriptional regulation and chromatin structure. In particular, we observed regional epigenetic changes of the transcription factor genes ZNF69, ZNF700 and ZNF763 as well as the HOXA3, HOXB3 and HOXD3 genes. A similar clustering of differential methylation was found in the CpG islands of the HIST1 genes suggesting effects on chromatin remodeling. Conclusions: The study shows that early established differential methylation in neural iPSC derivatives with T21 are associated with a set of genes relevant for DS brain development, providing a novel framework for further studies on epigenetic changes and transcriptional dysregulation during T21 neurogenesis. -
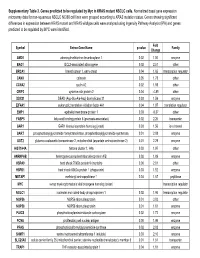
Supplementary Table 3. Genes Predicted to Be Regulated by Myc in KRAS Mutant NSCLC Cells
Supplementary Table 3. Genes predicted to be regulated by Myc in KRAS mutant NSCLC cells. Normalized basal gene expression microarray data for non-squamous NSCLC NCI60 cell lines were grouped according to KRAS mutation status. Genes showing significant differences in expression between KRAS mutant and KRAS wildtype cells were analyzed using Ingenuity Pathway Analysis (IPA) and genes predicted to be regulated by MYC were identified. Fold Symbol Entrez Gene Name p-value Family Change AMD1 adenosylmethionine decarboxylase 1 0.02 1.50 enzyme BAG1 BCL2-associated athanogene 0.02 2.57 other BRCA1 breast cancer 1, early onset 0.04 1.65 transcription regulator CANX calnexin 0.05 1.78 other CCNA2 cyclin A2 0.02 1.98 other CRIP2 cysteine-rich protein 2 0.04 -4.89 other DDX21 DEAD (Asp-Glu-Ala-Asp) box helicase 21 0.03 1.59 enzyme EIF4A1 eukaryotic translation initiation factor 4A1 0.04 1.87 translation regulator EMP1 epithelial membrane protein 1 0.03 -6.37 other FABP5 fatty acid binding protein 5 (psoriasis-associated) 0.02 2.26 transporter GAR1 GAR1 ribonucleoprotein homolog (yeast) 0.03 1.56 ion channel GART phosphoribosylglycinamide formyltransferase, phosphoribosylglycinamide synthetase, 0.01 2.08 enzyme GOT2 glutamic-oxaloacetic transaminase 2, mitochondrial (aspartate aminotransferase 2) 0.01 2.29 enzyme HIST1H4A histone cluster 1, H4a 0.03 1.97 other HNRNPAB heterogeneous nuclear ribonucleoprotein A/B 0.02 1.89 enzyme HSPA9 heat shock 70kDa protein 9 (mortalin) 0.00 2.01 other HSPD1 heat shock 60kDa protein 1 (chaperonin) 0.03 1.52 enzyme -

Identification of Differentially Expressed Genes in Human Bladder Cancer Through Genome-Wide Gene Expression Profiling
521-531 24/7/06 18:28 Page 521 ONCOLOGY REPORTS 16: 521-531, 2006 521 Identification of differentially expressed genes in human bladder cancer through genome-wide gene expression profiling KAZUMORI KAWAKAMI1,3, HIDEKI ENOKIDA1, TOKUSHI TACHIWADA1, TAKENARI GOTANDA1, KENGO TSUNEYOSHI1, HIROYUKI KUBO1, KENRYU NISHIYAMA1, MASAKI TAKIGUCHI2, MASAYUKI NAKAGAWA1 and NAOHIKO SEKI3 1Department of Urology, Graduate School of Medical and Dental Sciences, Kagoshima University, 8-35-1 Sakuragaoka, Kagoshima 890-8520; Departments of 2Biochemistry and Genetics, and 3Functional Genomics, Graduate School of Medicine, Chiba University, 1-8-1 Inohana, Chuo-ku, Chiba 260-8670, Japan Received February 15, 2006; Accepted April 27, 2006 Abstract. Large-scale gene expression profiling is an effective CKS2 gene not only as a potential biomarker for diagnosing, strategy for understanding the progression of bladder cancer but also for staging human BC. This is the first report (BC). The aim of this study was to identify genes that are demonstrating that CKS2 expression is strongly correlated expressed differently in the course of BC progression and to with the progression of human BC. establish new biomarkers for BC. Specimens from 21 patients with pathologically confirmed superficial (n=10) or Introduction invasive (n=11) BC and 4 normal bladder samples were studied; samples from 14 of the 21 BC samples were subjected Bladder cancer (BC) is among the 5 most common to microarray analysis. The validity of the microarray results malignancies worldwide, and the 2nd most common tumor of was verified by real-time RT-PCR. Of the 136 up-regulated the genitourinary tract and the 2nd most common cause of genes we detected, 21 were present in all 14 BCs examined death in patients with cancer of the urinary tract (1-7). -

Transcriptional and Post-Transcriptional Regulation of ATP-Binding Cassette Transporter Expression
Transcriptional and Post-transcriptional Regulation of ATP-binding Cassette Transporter Expression by Aparna Chhibber DISSERTATION Submitted in partial satisfaction of the requirements for the degree of DOCTOR OF PHILOSOPHY in Pharmaceutical Sciences and Pbarmacogenomies in the Copyright 2014 by Aparna Chhibber ii Acknowledgements First and foremost, I would like to thank my advisor, Dr. Deanna Kroetz. More than just a research advisor, Deanna has clearly made it a priority to guide her students to become better scientists, and I am grateful for the countless hours she has spent editing papers, developing presentations, discussing research, and so much more. I would not have made it this far without her support and guidance. My thesis committee has provided valuable advice through the years. Dr. Nadav Ahituv in particular has been a source of support from my first year in the graduate program as my academic advisor, qualifying exam committee chair, and finally thesis committee member. Dr. Kathy Giacomini graciously stepped in as a member of my thesis committee in my 3rd year, and Dr. Steven Brenner provided valuable input as thesis committee member in my 2nd year. My labmates over the past five years have been incredible colleagues and friends. Dr. Svetlana Markova first welcomed me into the lab and taught me numerous laboratory techniques, and has always been willing to act as a sounding board. Michael Martin has been my partner-in-crime in the lab from the beginning, and has made my days in lab fly by. Dr. Yingmei Lui has made the lab run smoothly, and has always been willing to jump in to help me at a moment’s notice. -

A Mutation in Histone H2B Represents a New Class of Oncogenic Driver
Author Manuscript Published OnlineFirst on July 23, 2019; DOI: 10.1158/2159-8290.CD-19-0393 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. A Mutation in Histone H2B Represents A New Class Of Oncogenic Driver Richard L. Bennett1, Aditya Bele1, Eliza C. Small2, Christine M. Will2, Behnam Nabet3, Jon A. Oyer2, Xiaoxiao Huang1,9, Rajarshi P. Ghosh4, Adrian T. Grzybowski5, Tao Yu6, Qiao Zhang7, Alberto Riva8, Tanmay P. Lele7, George C. Schatz9, Neil L. Kelleher9 Alexander J. Ruthenburg5, Jan Liphardt4 and Jonathan D. Licht1 * 1 Division of Hematology/Oncology, University of Florida Health Cancer Center, Gainesville, FL 2 Division of Hematology/Oncology, Northwestern University 3 Department of Cancer Biology, Dana Farber Cancer Institute and Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School 4 Department of Bioengineering, Stanford University 5 Department of Molecular Genetics and Cell Biology, The University of Chicago 6 Department of Chemistry, Tennessee Technological University 7 Department of Chemical Engineering, University of Florida 8 Bioinformatics Core, Interdisciplinary Center for Biotechnology Research, University of Florida 9 Department of Chemistry, Northwestern University, Evanston IL 60208 Running title: Histone mutations in cancer *Corresponding Author: Jonathan D. Licht, MD The University of Florida Health Cancer Center Cancer and Genetics Research Complex, Suite 145 2033 Mowry Road Gainesville, FL 32610 352-273-8143 [email protected] Disclosures: The authors have no conflicts of interest to declare Downloaded from cancerdiscovery.aacrjournals.org on September 27, 2021. © 2019 American Association for Cancer Research. Author Manuscript Published OnlineFirst on July 23, 2019; DOI: 10.1158/2159-8290.CD-19-0393 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. -

Human Recombinant Protein – TP760406
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for TP760406 H4C1 (NM_003538) Human Recombinant Protein Product data: Product Type: Recombinant Proteins Description: Purified recombinant protein of Human histone cluster 1, H4a (HIST1H4A), full length, with N- terminal HIS tag, expressed in E.Coli, 50ug Species: Human Expression Host: E. coli Tag: N-His Predicted MW: 11.2 kDa Concentration: >50 ug/mL as determined by microplate BCA method Purity: > 80% as determined by SDS-PAGE and Coomassie blue staining Buffer: 25mM Tris, pH8.0, 150 mM NaCl, 10% glycerol, 1 % Sarkosyl Storage: Store at -80°C. Stability: Stable for 12 months from the date of receipt of the product under proper storage and handling conditions. Avoid repeated freeze-thaw cycles. RefSeq: NP_003529 Locus ID: 8359 UniProt ID: P62805, B2R4R0 RefSeq Size: 372 Cytogenetics: 6p22.2 RefSeq ORF: 309 Synonyms: H4-16; H4C2; H4C3; H4C4; H4C5; H4C6; H4C8; H4C9; H4C11; H4C12; H4C13; H4C14; H4C15; H4FA; HIST1H4A This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 2 H4C1 (NM_003538) Human Recombinant Protein – TP760406 Summary: Histones are basic nuclear proteins that are responsible for the nucleosome structure of the chromosomal fiber in eukaryotes. Two molecules of each of the four core histones (H2A, H2B, H3, and H4) form an octamer, around which approximately 146 bp of DNA is wrapped in repeating units, called nucleosomes. -

Interindividual Differences in the Expression of ATP-Binding
Supplemental material to this article can be found at: http://dmd.aspetjournals.org/content/suppl/2018/02/02/dmd.117.079061.DC1 1521-009X/46/5/628–635$35.00 https://doi.org/10.1124/dmd.117.079061 DRUG METABOLISM AND DISPOSITION Drug Metab Dispos 46:628–635, May 2018 Copyright ª 2018 by The American Society for Pharmacology and Experimental Therapeutics Special Section on Transporters in Drug Disposition and Pharmacokinetic Prediction Interindividual Differences in the Expression of ATP-Binding Cassette and Solute Carrier Family Transporters in Human Skin: DNA Methylation Regulates Transcriptional Activity of the Human ABCC3 Gene s Tomoki Takechi, Takeshi Hirota, Tatsuya Sakai, Natsumi Maeda, Daisuke Kobayashi, and Ichiro Ieiri Downloaded from Department of Clinical Pharmacokinetics, Graduate School of Pharmaceutical Sciences, Kyushu University, Fukuoka, Japan (T.T., T.H., T.S., N.M., I.I.); Drug Development Research Laboratories, Kyoto R&D Center, Maruho Co., Ltd., Kyoto, Japan (T.T.); and Department of Clinical Pharmacy and Pharmaceutical Care, Graduate School of Pharmaceutical Sciences, Kyushu University, Fukuoka, Japan (D.K.) Received October 19, 2017; accepted January 30, 2018 dmd.aspetjournals.org ABSTRACT The identification of drug transporters expressed in human skin and levels. ABCC3 expression levels negatively correlated with the methylation interindividual differences in gene expression is important for understanding status of the CpG island (CGI) located approximately 10 kilobase pairs the role of drug transporters in human skin. In the present study, we upstream of ABCC3 (Rs: 20.323, P < 0.05). The reporter gene assay revealed evaluated the expression of ATP-binding cassette (ABC) and solute carrier a significant increase in transcriptional activity in the presence of CGI. -

University of California, San Diego
UC San Diego UC San Diego Electronic Theses and Dissertations Title The post-terminal differentiation fate of RNAs revealed by next-generation sequencing Permalink https://escholarship.org/uc/item/7324r1rj Author Lefkowitz, Gloria Kuo Publication Date 2012 Peer reviewed|Thesis/dissertation eScholarship.org Powered by the California Digital Library University of California UNIVERSITY OF CALIFORNIA, SAN DIEGO The post-terminal differentiation fate of RNAs revealed by next-generation sequencing A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Biomedical Sciences by Gloria Kuo Lefkowitz Committee in Charge: Professor Benjamin D. Yu, Chair Professor Richard Gallo Professor Bruce A. Hamilton Professor Miles F. Wilkinson Professor Eugene Yeo 2012 Copyright Gloria Kuo Lefkowitz, 2012 All rights reserved. The Dissertation of Gloria Kuo Lefkowitz is approved, and it is acceptable in quality and form for publication on microfilm and electronically: __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ Chair University of California, San Diego 2012 iii DEDICATION Ma and Ba, for your early indulgence and support. Matt and James, for choosing more practical callings. Roy, my love, for patiently sharing the ups and downs -

ABCC11 Antibody (F48871)
ABCC11 Antibody (F48871) Catalog No. Formulation Size F48871-0.4ML In 1X PBS, pH 7.4, with 0.09% sodium azide 0.4 ml F48871-0.08ML In 1X PBS, pH 7.4, with 0.09% sodium azide 0.08 ml Bulk quote request Availability 1-3 business days Species Reactivity Human Format Antigen affinity purified Clonality Polyclonal (rabbit origin) Isotype Rabbit Ig Purity Antigen affinity UniProt Q96J66 Localization Cytoplasmic, membranous Applications Western blot : 1:1000 IHC (Paraffin) : 1:50-1:100 Limitations This ABCC11 antibody is available for research use only. ABCC11 antibody IHC analysis in formalin fixed and paraffin embedded breast carcinoma. Western blot analysis of ABCC11 antibody and WiDr lysate. Predicted molecular weight: ~154/150kDa (isoforms 1/2). Description ATP-binding cassette sub-family C member 11 is a member of the superfamily of ATP-binding cassette (ABC) transporters. ABC proteins transport various molecules across extra- and intra-cellular membranes. ABC genes are divided into seven distinct subfamilies (ABC1, MDR/TAP, MRP, ALD, OABP, GCN20, White). This ABC full transporter is a member of the MRP subfamily which is involved in multi-drug resistance. The product of this gene participates in physiological processes involving bile acids, conjugated steroids, and cyclic nucleotides. In addition, a SNP in this gene is responsible for determination of human earwax type. This gene and family member ABCC12 are determined to be derived by duplication and are both localized to chromosome 16q12.1. Application Notes Titration of the ABCC11 antibody may be required due to differences in protocols and secondary/substrate sensitivity. Immunogen A portion of amino acids 343-372 from the human protein was used as the immunogen for this ABCC11 antibody. -

Acetyl-Histone H4-K12 Pab Cat
Acetyl-Histone H4-K12 pAb Cat. No.: 15-437 Acetyl-Histone H4-K12 pAb Immunohistochemistry of paraffin- embedded rat ovary using Acetyl-Histone Immunohistochemistry of paraffin-embedded mouse testis using H4-K12 antibody (15-437) at dilution of 1:200 Acetyl-Histone H4-K12 antibody (15-437) at dilution of 1:200 (40x lens). (40x lens). Immunohistochemistry of paraffin- Immunofluorescence analysis of C6 cells using Acetyl-Histone H4-K12 embedded mouse spinal cord using Acetyl- antibody (15-437) at dilution of 1:100.C6 cells were treated by TSA (1 Histone H4-K12 antibody (15-437) at dilution uM) at 37? for 18 hours. Blue: DAPI for nuclear staining. of 1:200 (40x lens). September 27, 2021 1 https://www.prosci-inc.com/acetyl-histone-h4-k12-pab-15-437.html Immunofluorescence analysis of NIH/3T3 cells using Acetyl-Histone H4-K12 antibody (15-437) at dilution of 1:100.NIH/3T3 cells were treated by TSA (1 uM) at 37? for 18 hours. Blue: DAPI for nuclear staining. Specifications HOST SPECIES: Rabbit SPECIES REACTIVITY: Human, Mouse IMMUNOGEN: A synthetic acetylated peptide around K12 of human Histone H4 (NP_001029249.1). TESTED APPLICATIONS: IF, IHC, WB WB: ,1:500 - 1:2000 APPLICATIONS: IHC: ,1:50 - 1:200 IF: ,1:50 - 1:200 Properties PURIFICATION: Affinity purification CLONALITY: Polyclonal ISOTYPE: IgG CONJUGATE: Unconjugated PHYSICAL STATE: Liquid BUFFER: PBS with 0.02% sodium azide, 50% glycerol, pH7.3. STORAGE CONDITIONS: Store at -20˚C. Avoid freeze / thaw cycles. Additional Info OFFICIAL SYMBOL: H4K12ac September 27, 2021 2 https://www.prosci-inc.com/acetyl-histone-h4-k12-pab-15-437.html H4/A, H4FA, HIST1H4B, H4/I, H4FI, HIST1H4C, H4/G, H4FG, HIST1H4D, H4/B, H4FB, HIST1H4E, H4/J, H4FJ, HIST1H4F, H4/C, H4FC, HIST1H4H, H4/H, H4FH, HIST1H4I, H4/M, ALTERNATE NAMES: H4FM, HIST1H4J, H4/E, H4FE, HIST1H4K, H4/D, H4FD, HIST1H4L, H4/K, H4FK, HIST2H4A, H4/N, H4F2, H4FN, HIST2H4, HIST2H4B, H4/O, H4FO, HIST4H4, Histone H4H4FA GENE ID: 8366 USER NOTE: Optimal dilutions for each application to be determined by the researcher.