RNA Synthesis by in Vitro Selected Ribozymes for Recreating an RNA World
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Intelligent Design, Abiogenesis, and Learning from History: Dennis R
Author Exchange Intelligent Design, Abiogenesis, and Learning from History: Dennis R. Venema A Reply to Meyer Dennis R. Venema Weizsäcker’s book The World View of Physics is still keeping me very busy. It has again brought home to me quite clearly how wrong it is to use God as a stop-gap for the incompleteness of our knowledge. If in fact the frontiers of knowledge are being pushed back (and that is bound to be the case), then God is being pushed back with them, and is therefore continually in retreat. We are to find God in what we know, not in what we don’t know; God wants us to realize his presence, not in unsolved problems but in those that are solved. Dietrich Bonhoeffer1 am thankful for this opportunity to nature, is the result of intelligence. More- reply to Stephen Meyer’s criticisms over, this assertion is proffered as the I 2 of my review of his book Signature logical basis for inferring design for the in the Cell (hereafter Signature). Meyer’s origin of biological information: if infor- critiques of my review fall into two gen- mation only ever arises from intelli- eral categories. First, he claims I mistook gence, then the mere presence of Signature for an argument against bio- information demonstrates design. A few logical evolution, rendering several of examples from Signature make the point my arguments superfluous. Secondly, easily: Meyer asserts that I have failed to refute … historical scientists can show that his thesis by not providing a “causally a presently acting cause must have adequate alternative explanation” for the been present in the past because the origin of life in that the few relevant cri- proposed candidate is the only known tiques I do provide are “deeply flawed.” cause of the effect in question. -

The Hidden Kingdom
INTRODUCTION Fungi—The Hidden Kingdom OBJECTIVE • To provide students with basic knowledge about fungi Activity 0.1 BACKGROUND INFORMATION The following text provides an introduction to the fungi. It is written with the intention of sparking curiosity about this GRADES fascinating biological kingdom. 4-6 with a K-3 adaptation TEACHER INSTRUCTIONS TYPE OF ACTIVITY 1. With your class, brainstorm everything you know about fungi. Teacher read/comprehension 2. For younger students, hand out the question sheet before you begin the teacher read and have them follow along and MATERIALS answer the questions as you read. • copies of page 11 3. For older students, inform them that they will be given a • pencils brainteaser quiz (that is not for evaluation) after you finish reading the text. VOCABULARY 4. The class can work on the questions with partners or in groups bioremediation and then go over the answers as a class. Discuss any chitin particularly interesting facts and encourage further fungi independent research. habitat hyphae K-3 ADAPTATION kingdom 1. To introduce younger students to fungi, you can make a KWL lichens chart either as a class or individually. A KWL chart is divided moulds into three parts. The first tells what a student KNOWS (K) mushrooms about a subject before it is studied in class. The second part mycelium tells what the student WANTS (W) to know about that subject. mycorrhizas The third part tells what the child LEARNED (L) after studying nematodes that subject. parasitic fungi 2. Share some of the fascinating fungal facts presented in the photosynthesis “Fungi—The Hidden Kingdom” text with your students. -

Framing Major Prebiotic Transitions As Stages of Protocell Development: Three Challenges for Origins-Of-Life Research
Framing major prebiotic transitions as stages of protocell development: three challenges for origins-of-life research Ben Shirt-Ediss1, Sara Murillo-Sánchez2,3 and Kepa Ruiz-Mirazo*2,3 Commentary Open Access Address: Beilstein J. Org. Chem. 2017, 13, 1388–1395. 1Interdisciplinary Computing and Complex BioSystems Group, doi:10.3762/bjoc.13.135 University of Newcastle, UK, 2Dept. Logic and Philosophy of Science, University of the Basque Country, Spain and 3Biofisika Institute Received: 16 February 2017 (CSIC, UPV-EHU), Spain Accepted: 27 June 2017 Published: 13 July 2017 Email: Kepa Ruiz-Mirazo* - [email protected] This article is part of the Thematic Series "From prebiotic chemistry to molecular evolution". * Corresponding author Guest Editor: L. Cronin Keywords: functional integration; origins of life; prebiotic evolution; protocells © 2017 Shirt-Ediss et al.; licensee Beilstein-Institut. License and terms: see end of document. Abstract Conceiving the process of biogenesis as the evolutionary development of highly dynamic and integrated protocell populations provides the most appropriate framework to address the difficult problem of how prebiotic chemistry bridged the gap to full-fledged living organisms on the early Earth. In this contribution we briefly discuss the implications of taking dynamic, functionally inte- grated protocell systems (rather than complex reaction networks in bulk solution, sets of artificially evolvable replicating molecules, or even these same replicating molecules encapsulated in passive compartments) -

The Concept of Organism, a Historical and Conceptual Critique
Do organisms have an ontological status? Charles T. Wolfe Unit for History and Philosophy of Science University of Sydney [email protected] forthcoming in History and Philosophy of the Life Sciences 32:2-3 (2010) Abstract The category of „organism‟ has an ambiguous status: is it scientific or is it philosophical? Or, if one looks at it from within the relatively recent field or sub-field of philosophy of biology, is it a central, or at least legitimate category therein, or should it be dispensed with? In any case, it has long served as a kind of scientific “bolstering” for a philosophical train of argument which seeks to refute the “mechanistic” or “reductionist” trend, which has been perceived as dominant since the 17th century, whether in the case of Stahlian animism, Leibnizian monadology, the neo-vitalism of Hans Driesch, or, lastly, of the “phenomenology of organic life” in the 20th century, with authors such as Kurt Goldstein, Maurice Merleau-Ponty, and Georges Canguilhem. In this paper I try to reconstruct some of the main interpretive „stages‟ or „layers‟ of the concept of organism in order to critically evaluate it. How might „organism‟ be a useful concept if one rules out the excesses of „organismic‟ biology and metaphysics? Varieties of instrumentalism and what I call the „projective‟ concept of organism are appealing, but perhaps ultimately unsatisfying. 1. What is an organism? There have been a variety of answers to this question, not just in the sense of different definitions (an organism is a biological individual; it is a living being, or at least the difference between a living organism and a dead organism is somehow significant in a way that does not seem to make sense for other sorts of entities, like lamps and chairs; it is a self-organizing, metabolic system; etc.) but more tendentiously, in the 1 sense that philosophers, scientists, „natural philosophers‟ and others have both asserted and denied the existence of organisms. -

Bacteria – Friend Or Foe?
Bacteria – Friend or Foe? By Rachel L. Dittmar & Rosa M. Santana Carrero [email protected], [email protected] April 2020 Danger! Sick! Gross! Vaccine! Disease! Medicine! Dirty! MRSA! Eww! E. coli! What is the first thing that comes to mind when you hear “bacteria”? Science! Microscope! MRSA! Salmonella! Hospital! Germs! Staph! Food! Small! 2 Introduction to Bacteria 3 Bacterial nomenclature • Bacteria are referred to by their genus and species, with genus coming first and species coming last: Escherichia coli Escherichia: genus Species: coli Bacteria names are ALWAYS italicized. Genus names are capitalized and species names are not. Sometimes, the genus is abbreviated by its first initial: E. coli 4 Bacteria are prokaryotes. 5 What are prokaryotes? • Plasma membrane separates the cell from its surrounding environment • Cytoplasm contains organelles • Contain DNA consisting of a single large, circular chromosome • Ribosomes make proteins 6 Prokaryotes v. Eukaryotes 7 Image from: https://www.difference.wiki/prokaryotic-cell-vs-eukaryotic-cell/ How do these organisms differ? Prokaryotes Eukaryotes - circular DNA - linear DNA (found in nucleus) - no nucleus - nucleus - no membrane bound organelles - membrane bound organelles - small- less than 10μm - larger than 10μm - unicellular - can be unicellular and multicellular 8 Bacteria are very small. 9 How big are bacteria? Bacteria are very small: 0.1 – 5.0 micrometers. A micrometer (μm) is 0.000001 meters or 0.001 millimeters (mm) For comparison, a human hair is 30 – 100 μm Image from: https://www.khanacademy.org/science/high-school-biology/hs-cells/hs-prokaryotes-and-eukaryotes/a/prokaryotic-cells 10 Bacteria are classified by phenotype or genotype. -

Single-Celled Organisms
Single-Celled Organisms • Humans are made up of millions of tiny cells (multi-celled organisms). • Microorganisms are made up of only ONE cell (single-celled organisms) 1.Single-celled organisms are simple life forms Include: bacteria, protists and others. They move, find food, grow, and make new organisms. 2. Bacteria are one-celled organisms that do NOT have a nucleus. The material that is usually in a nucleus can be found all over the cytoplasm instead. More bacteria on earth than any other living thing Some bacteria are helpful, and some are harmful. One handful of soil can contain billions of bacteria. 3. Protists are a kind of single-celled organism. They live everywhere including pond water, saltwater, animals, and humans Amoebas have a cell membrane, cytoplasm, and a nucleus They move by: changing their shape. 4. Algae are plant-like organisms that Live in water, moist places, and under rocks Among the oldest things on earth! Algae have a cell membrane, cytoplasm, nucleus, and chloroplast. Algae produce most of the oxygen that you breathe! 5. How do single-celled organisms move? Tail-like parts called flagella Hair like structures called cilia Paramecium also use their cilia to get food Multi-Celled Organisms Multi-celled organism is made of up of more than one cell. Plants, animals and other living things. Made up of different kinds of cells that have certain jobs to do. Muscle cells, white blood cells, and nerve cells For example: the cells that make up your heart work together to move oxygen and blood to other cells throughout your body. -

Biology 197: Principles of Organismal Biology Section B: T, R 9:30–10:45 AM Section D: T, R 1:30–2:45 PM Spring 2011 Birck Hall 003 Instructor: Dr
Biology 197: Principles of Organismal Biology Section B: T, R 9:30–10:45 AM Section D: T, R 1:30–2:45 PM Spring 2011 Birck Hall 003 Instructor: Dr. Phil Novack‐Gottshall Office: Birck 332 E‐mail: Blackboard mail preferred Office hours: T 11–1:30, W 11–1, R 11–12:30 (or pnovack‐[email protected]) or by appointment Course Description Organismal biology is one of the major branches of biology and is concerned with all aspects of the life of organisms, including their biodiversity, anatomical structure, physiology, development, biogeography, and ecology. This course is an introductory course required for all biological sciences majors, but it is also useful for gaining basic biological literacy and for those pursuing careers in human and veterinary medicine, psychiatry, agriculture, forestry, microbiology, conservation, ecology, paleontology, environmental science, law, political science, and even cooking, cheese making, and brewing of alcohol. In this class, we will learn the major groups of animals, fungi, plants, protists, algae, and bacteria; their basic characteristics; and how biologists study these organisms to understand their rich evolutionary history, ecological interactions, amazing adaptations, and relevance to humans and other species. In particular, you will practice learning how to view the world and to think like an organismal biologist. Learning objectives 1) Explain the scientific method of organismal biologists to understand the natural world. 2) Identify the major lineages of life through study of their biodiversity, anatomy, physiology, development, behavior, biogeography, fossil record, and ecology. 3) Explain the significance of major transitions in organismal evolution: photosynthesis, endosymbiosis, sexual reproduction, multicellularity, skeletonization, cephalization, terrestrialization, mobility, and carnivory, among others. -

Quantifying the Origins of Life on a Planetary Scale
Quantifying the origins of life on a planetary scale Caleb Scharfa,1 and Leroy Croninb,1 aColumbia Astrobiology Center, Columbia Astrophysics Laboratory, New York, NY 10027; and bSchool of Chemistry, University of Glasgow, Glasgow, G12 8QQ, United Kingdom Edited by Neta A. Bahcall, Princeton University, Princeton, NJ, and approved May 17, 2016 (received for review November 23, 2015) A simple, heuristic formula with parallels to the Drake Equation is currently impossible to construct a first principles quantitative introduced to help focus discussion on open questions for the origins estimate of an abiogenesis probability for the young Earth. It is of life in a planetary context. This approach indicates a number of also the case that we typically conflate the idea of “microabio- areas where quantitative progress can be made on parameter genesis”— namely, a highly localized assembly of molecular estimation for determining origins of life probabilities, based on structures that meets the minimum criterion for a living system— constraints from Bayesian approaches. We discuss a variety of “micro- with “macro-” or planet-wide abiogenesis. In other words the idea scale” factors and their role in determining “macroscale” abiogenesis of life arising on a planet is treated as a singular event rather than probabilities on suitable planets. We also propose that impact ejecta a possible accumulation of many microscale events, each of which exchange between planets with parallel chemistries and chemical evo- might be considered an origin event in its own right, and are ar- lution could in principle amplify the development of molecular com- guably more accessible through laboratory or modeling investi- plexity and abiogenesis probabilities. -

Chapter 1: the Human Organism 1 Chapter 1 Outline 1.1 Anatomy and Physiology 1.2 Structural and Functional Organization of the Human Body A
Chapter 1 The Human Organism Chapter 1: The Human Organism 1 Chapter 1 Outline 1.1 Anatomy and Physiology 1.2 Structural and functional organization of the human body A. 11 Organ Systems 1.3 Characteristics of Life 1.4 Biomedical Research 1.5 Homeostasis A. Negative feedback B. Positive feedback 1.6 Terminology and the Body plan A. Body Position B. Directional terms C. Body parts and regions D. Planes E. Body Cavities F. Serous Membranes Chapter 1: The Human Organism 2 1.1 Anatomy and Physiology Anatomy Physiology • Scientific discipline that • Scientific investigation of investigates body the processes or functions of living things. structure & examines the • Goal: relationship between – Understand & predict body’s structure and function responses to stimuli – Understand how the body maintains conditions with a narrow range of values in a constantly changing environment. Study of the human body encompasses both because they are highly interwoven. Chapter 1: The Human Organism 3 Various types of study: • Anatomy: • Physiology: – Developmental Anatomy – Cell Physiology • Embryology – Systemic Physiology – Cytology – Neurophysiology • Histology – Cardiovascular – Gross Anatomy physiology • Regional – Exercise physiology • Systemic Usually physiological – Surface Anatomy study is systemic – Anatomical Anomalies because functions occur in multiple places in the body. – Pathology Chapter 1: The Human Organism 4 Anatomical Imaging 1. Radiograph 2. Ultrasound 3. Computed tomography 4. Dynamic subtraction angiography 5. Magnetic resonance imaging 6. Positron emission tomography • Table 1.1 Page 3 Chapter 1: The Human Organism 5 1.2 Struc & Fxnl Organization- Human Body 1. Chemical Level: Involves interactions of atoms coming 2. Cellular Level: together to form more Basic structural & 3. -

The RNA World: a Critique
The RNA World: A Critique - Origins & Design 17:1. Mills, Gordon and... http://www.arn.org/docs/odesign/od171/rnaworld171.htm Access Research Network Origins & Design Archives Review Article Origins & Design 17:1 The RNA World: A Critique Gordon C. Mills Department of Human Biological Chemistry and Genetics University of Texas Medical Branch Galveston, TX 77555 Dean Kenyon Department of Biology San Francisco State University 1600 Holloway Avenue San Francisco, CA 94132 Introduction One of the earliest published suggestions that RNA-catalyzed RNA replication preceded and gave rise to the first DNA-based living cells was made by Carl Woese in 1967, in his book The Genetic Code1. Similar suggestions were made by Crick and Orgel2, for reasons that are not difficult to grasp. Prior to the discovery of catalytic RNAs, proteins were considered by many to be the only organic molecules in living matter that could function as catalysts. DNA carries the genetic information required for the synthesis of proteins. The replication and transcription of DNA require a complex set of enzymes and other proteins. How then could the first living cells with DNA-based molecular biology have originated by spontaneous chemical processes on the prebiotic Earth? Primordial DNA synthesis would have required the presence of specific enzymes, but how could these enzymes be synthesized without the genetic information in DNA and without RNA for translating that information into the amino acid sequence of the protein enzymes? In other words, proteins are required for DNA synthesis and DNA is required for protein synthesis. This classic "chicken-and-egg" problem made it immensely difficult to conceive of any plausible prebiotic chemical pathway to the molecular biological system. -

Cooperative Evolution Reclaiming Darwin’S Vision
COOPERATIVE EVOLUTION RECLAIMING DARWIN’S VISION COOPERATIVE EVOLUTION RECLAIMING DARWIN’S VISION CHRISTOPHER BRYANT AND VALERIE A. BROWN For Annie Bryant who has had to put up with parasites and mitochondria at the dinner table for 58 years and, in spite of that, managed to pass on to our children a very healthy mitochondrial genome, with love. And for Chris, Sarah, Elliot and Amon Brown who are taking the next steps in cooperative evolution. By mutual confidence and mutual aid, Great deeds are done and great discoveries made. – Homer Published by ANU Press The Australian National University Acton ACT 2601, Australia Email: [email protected] Available to download for free at press.anu.edu.au ISBN (print): 9781760464288 ISBN (online): 9781760464295 WorldCat (print): 1240754622 WorldCat (online): 1240754606 DOI: 10.22459/CE.2021 This title is published under a Creative Commons Attribution-NonCommercial- NoDerivatives 4.0 International (CC BY-NC-ND 4.0). The full licence terms are available at creativecommons.org/licenses/by-nc-nd/4.0/legalcode Cover design and layout by ANU Press This edition © 2021 ANU Press CONTENTS Acknowledgements . xi Glossary of Words and Phrases . xiii Introduction . 1 1 . In Homage to Darwin . 5 2 . All Knowledge is Metaphor . 17 3 . Intelligent Evolution and Intelligence . 31 4 . How Evolution Works . 55 5 . The Past is a Foreign Country . 75 6 . We Do Things Differently Now . 89 7 . Energy: Where it all Begins . 103 8 . Everything is Connected . 119 9 . Walling In and Walling Out . 133 10 . Becoming Human . 149 11 . Inheriting the Earth . 163 12 . Our Closest Cousins . -
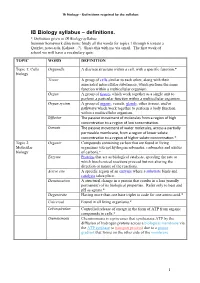
IB Biology Syllabus – Definitions
IB Biology – Definitions required by the syllabus IB Biology syllabus – definitions. * Definition given in IB Biology syllabus Summer homework directions: Study all the words for topics 1 through 6 (create a Quizlet, notecards, Kahoot…?). Share this with me via email. The first week of school we will have a vocabulary quiz. TOPIC WORD DEFINITION Topic 1: Cells Organelle A discrete structure within a cell, with a specific function.* biology Tissue A group of cells similar to each other, along with their associated intercellular substances, which perform the same function within a multicellular organism. Organ A group of tissues which work together as a single unit to perform a particular function within a multicellular organism. Organ system A group of organs, vessels, glands, other tissues, and/or pathways which work together to perform a body function within a multicellular organism. Diffusion The passive movement of molecules from a region of high concentration to a region of low concentration. Osmosis The passive movement of water molecules, across a partially permeable membrane, from a region of lower solute concentration to a region of higher solute concentration.* Topic 2: Organic Compounds containing carbon that are found in living Molecular organisms (except hydrogencarbonates, carbonates and oxides biology of carbon).* Enzyme Proteins that act as biological catalysts, speeding the rate at which biochemical reactions proceed but not altering the direction or nature of the reactions. Active site A specific region of an enzyme where a substrate binds and catalysis takes place. Denaturation A structural change in a protein that results in a loss (usually permanent) of its biological properties.