Oxygen Isotopic Composition of Ferric Oxides from Recent Soil, Hydrologic, and Marine Environments
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Bedrock Geology of Dodge County, Wisconsin (Wisconsin Geological
MAP 508 • 2021 Bedrock geology of Dodge County, Wisconsin DODGE COUNTY Esther K. Stewart 88°30' 88°45' 88°37'30" 88°52'30" 6 EXPLANATION OF MAP UNITS Tunnel City Group, undivided (Furongian; 0–155 ft) FOND DU LAC CO 630 40 89°0' 6 ! 6 20 ! 10 !! ! ! A W ! ! 1100 W ! GREEN LAKE CO ! ! ! WW ! ! ! ! DG-92 ! ! ! 1100 B W! Includes Lone Rock and Mazomanie Formations. These formations are both DG-53 W ! «49 ! CORRELATION OF MAP UNITS !! ! 7 ! !W ! ! 43°37'30" R16E _tc EL709 DG-1205 R15E W R14E R15E DG-24 W! ! 1 Quaternary ! 980 ! W W 1 ! ! ! 6 DG-34 6 _ ! 1 R17E Os Lake 1 R16E 6 interbedded and laterally discontinuous and therefore cannot be mapped 1 6 W ! ! 1100 !! 175 940 Waupun DG-51 ! 980 « Oa ! R13E 6 Emily R14E W ! 43°37'30" ! ! ! 41 ¤151 B «49 ! ! ! ! Opc ! Drew «68 ! W ! East ! ! ! individually at this scale in Dodge County. Overlies Elk Mound Group across KW313 940 ! ! ! ! ! ! 940 ! W B ! ! - ! ! W ! ! ! ! ! ! !! Waupun ! W ! Undifferentiated sediment ! ! W! B 000m Cr W! ! º Libby Cr ! 3 INTRUSIVE SUPRACRUSTAL 3 1020 ! ! Waupun ! DG-37 W ! ! º 1020 a sharp contact. W ! 50 50 N ! ! KS450 ! ! ! IG300 ! B B Airport ! RO703 ! ! Brownsville ! ! ! ! ! ! 1060 ! ROCKS W ! ! ROCKS Unconsolidated sediments deposited by modern and glacial processes. 940 ° ! Qu ! W Br Rock SQ463 B ! Pink, gray, white, and green; coarse- to fine-grained; moderately to poorly 980 B River B B ! ! KT383 ! ! Generally 20–60 feet (ft) thick; ranges from absent where bedrock crops ! !! ! ! ! ! ! Su Lower Silurian ° ! ! ! ! ! 940 860 ! ! ! ! ! ! ! ! ! ! sorted; glauconitic sandstone, siltstone, and mudstone with variable W ! B B B ! ! ! 980 ! ! ! 780 ! Kummel !! out to more than 200 ft thick in preglacial bedrock valleys. -

Petrology and Diagenesis of the Cyclic Maquoketa Formation (Upper Ordovician) Pike County, Missouri
Scholars' Mine Masters Theses Student Theses and Dissertations 1970 Petrology and diagenesis of the cyclic Maquoketa Formation (Upper Ordovician) Pike County, Missouri Edwin Carl Kettenbrink Jr. Follow this and additional works at: https://scholarsmine.mst.edu/masters_theses Part of the Geology Commons Department: Recommended Citation Kettenbrink, Edwin Carl Jr., "Petrology and diagenesis of the cyclic Maquoketa Formation (Upper Ordovician) Pike County, Missouri" (1970). Masters Theses. 5514. https://scholarsmine.mst.edu/masters_theses/5514 This thesis is brought to you by Scholars' Mine, a service of the Missouri S&T Library and Learning Resources. This work is protected by U. S. Copyright Law. Unauthorized use including reproduction for redistribution requires the permission of the copyright holder. For more information, please contact [email protected]. .,!) l PETROLOGY AND DIAGENESIS OF THE CYCLIC MAQUOKETA FORMATION (UPPER ORDOVICIAN) PIKE COUNTY, MISSOURI BY EDWIN CARL KETTENBRINK, JR., 1944- A THESIS submitted to the faculty of UNIVERSITY OF MISSOURI - ROLLA in partial fulfillment of the requirements for the Degree of MASTER OF SCIENCE IN GEOLOGY Rolla, Missouri 1970 T2532 231 pages c. I Approved by ~ (advisor) ~lo/ ii ABSTRACT The Maquoketa Formation (Upper Ordovician-Cincinnatian Series) has been extensively studied for over one hundred years, but a petrographic study of its cyclic lithologies has been neglected. The following six distinct Maquoketa lithologies have been recognized in this study in Pike County, Missouri& 1) phosphatic biosparite, 2) phos phorite, 3) micrite-microsparite, 4) dolomitic shale, 5) dolomitic marlstone, and 6) dolomitic quartz siltstone. Three cycle types are present in the Maquoketa. They are expressed as thin beds (1-20 inches) of alternating micrite-shale, micrite-marlstone, and shale-siltstone. -

Paleozoic Stratigraphic Nomenclature for Wisconsin (Wisconsin
UNIVERSITY EXTENSION The University of Wisconsin Geological and Natural History Survey Information Circular Number 8 Paleozoic Stratigraphic Nomenclature For Wisconsin By Meredith E. Ostrom"'" INTRODUCTION The Paleozoic stratigraphic nomenclature shown in the Oronto a Precambrian age and selected the basal contact column is a part of a broad program of the Wisconsin at the top of the uppermost volcanic bed. It is now known Geological and Natural History Survey to re-examine the that the Oronto is unconformable with older rocks in some Paleozoic rocks of Wisconsin and is a response to the needs areas as for example at Fond du Lac, Minnesota, where of geologists, hydrologists and the mineral industry. The the Outer Conglomerate and Nonesuch Shale are missing column was preceded by studies of pre-Cincinnatian cyclical and the younger Freda Sandstone rests on the Thompson sedimentation in the upper Mississippi valley area (Ostrom, Slate (Raasch, 1950; Goldich et ai, 1961). An unconformity 1964), Cambro-Ordovician stratigraphy of southwestern at the upper contact in the Upper Peninsula of Michigan Wisconsin (Ostrom, 1965) and Cambrian stratigraphy in has been postulated by Hamblin (1961) and in northwestern western Wisconsin (Ostrom, 1966). Wisconsin wlle're Atwater and Clement (1935) describe un A major problem of correlation is the tracing of outcrop conformities between flat-lying quartz sandstone (either formations into the subsurface. Outcrop definitions of Mt. Simon, Bayfield, or Hinckley) and older westward formations based chiefly on paleontology can rarely, if dipping Keweenawan volcanics and arkosic sandstone. ever, be extended into the subsurface of Wisconsin because From the above data it would appear that arkosic fossils are usually scarce or absent and their fragments cari rocks of the Oronto Group are unconformable with both seldom be recognized in drill cuttings. -

Upper Ordovician) at Wequiock Creek, Eastern Wisconsin
~rnooij~~~mij~rnoo~ ~oorn~rn~rn~~ rnoo~~rnrn~rn~~ Number 35 September, 1980 Stratigraphy and Paleontology of the Maquoketa Group (Upper Ordovician) at Wequiock Creek, Eastern Wisconsin Paul A. Sivon Department of Geology University of Illinois Urbana, Illinois REVIEW COMMITTEE FOR THIS CONTRIBUTION: T.N. Bayer, Winona State College University, Winona Minnesota M.E. Ostrom, Wisconsin Geological Survey, Madison, Wisconsin Peter Sheehan, Milwaukee Public Museum· ISBN :0-89326-016-4 Milwaukee Public Museum Press Published by the Order of the Board of Trustees Milwaukee Public Museum Accepted for publication July, 1980 Stratigraphy and Paleontology of the Maquoketa Group (Upper Ordovician) at Wequiock Creek, Eastern Wisconsin Paul A. Sivon Department of Geology University of Illinois Urbana, Illinois Abstract: The Maquoketa Group (Upper Ordovician) is poorly exposed in eastern Wisconsin. The most extensive exposure is found along Wequiock Creek, about 10 kilometers north of Green Bay. There the selection includes a small part of the upper Scales Shale and good exposures of the Fort Atkinson Limestone and Brainard Shale. The exposed Scales Shale is 2.4 m of clay, uniform in appearance and containing no apparent fossils. Limestone and dolomite dominate the 3.9 m thick Fort Atkinson Limestone. The carbonate beds alternate with layers of dolomitic shale that contain little to no fauna. The shales represent times of peak terrigenous clastic deposition in a quiet water environment. The car- bonates are predominantly biogenic dolomite and biomicrite. Biotic succession within single carbonate beds includes replacement of a strophomenid-Lepidocyclus dominated bottom community by a trep- ostome bryozoan-Plaesiomys-Lepidocyclus dominated community. Transported echinoderm and cryptostome bryozoan biocalcarenites are common. -

Paleozoic Lithostratigraphic Nomenclature for Minnesota
MINNESOTA GEOLOGICAL SURVEY PRISCILLA C. GREW, Director PALEOZOIC LITHOSTRATIGRAPHIC NOMENCLATURE FOR MINNESOTA John H. Mossier Report of Investigations 36 ISSN 0076-9177 UNIVERSITY OF MINNESOTA Saint Paul - 1987 PALEOZOIC LITHOSTRATIGRAPHIC NOMENCLATURE FOR MINNESOTA CONTENTS Abstract. Structural and sedimentological framework • Cambrian System • 2 Mt. Simon Sandstone. 2 Eau Claire Formation • 6 Galesville Sandstone • 8 Ironton Sandstone. 9 Franconia Formation. 9 St. Lawrence Formation. 11 Jordan Standstone. 12 Ordovician System. 13 Prairie du Chien Group. 14 Oneota Dolomite. 14 Shakopee Formation. 15 St. Peter Sandstone. 17 Glenwood Formation. 17 Platteville Formation. 18 Decorah Shale. 19 Galena Group • 22 Cummings ville Formation. 22 Prosser Limestone. 23 Stewartville Formation • 24 Dubuque Formation. 24 Maquoketa Formation. 25 Devonian System • 25 Spillville Formation • 26 Wapsipinicon Formation 26 Cedar Valley Formation • 26 Northwestern Minnesota. 28 Winnipeg Formation • 28 Red River Formation. 29 Acknowledgments • 30 References cited. 30 Appendix--Principal gamma logs used to construct the composite gamma log illustrated on Plate 1. 36 ILLUSTRATIONS Plate 1 • Paleozoic lithostratigraphic nomenclature for Minnesota • .in pocket Figure 1. Paleogeographic maps of southeastern Minnesota • 3 2. Map showing locations of outcrops, type sections, and cores, southeastern t1innesota • 4 3. Upper Cambrian stratigraphic nomenclature 7 iii Figure 4. Lower Ordovician stratigraphic nomenclature • • • • 14 5. Upper Ordovician stratigraphic nomenclature 20 6. Middle Devonian stratigraphic nomenclature. • • . • • 27 7. Map showing locations of cores and cuttings in northwestern Minnesota • • • • • • • • • • • • • • • • • • 29 TABLE Table 1. Representative cores in Upper Cambrian formations •••••• 5 The University of Minnesota is committed to the policy that all persons shall have equal access to its programs, facilities, and employment without regard to race, religion, color, sex, national orgin, handicap, age, veteran status, or sexual orientation. -

Study of Mined Storage Caverns
ORNL/SUB-75/64509 STUDY OF MINED STORAGE CAVERNS James H. Cobbs Enyinecring Tulsa, Oklahoma June, 1975 s SIOIICE V ability. pfjpT ! 7'-is This report was prepared by James H. Cobbs Engineering under Purchase Order Subcontract 78X-64509V with Union Carbide Corporation, Nuclear Division. The subcontract was adminis- tered by Oak Ridge National Laboratory v/ ' ' ."- . ,-r • OFFICE ,OF WASTE ISOLATION . i . '' . : L iui.-un - \P'A I, K RIDGE. TENN'ES.SEE* .!»•». • > v... prepared tor the U.S. ENERGY RESEARCH AND DEVELOPMENT ADMINISTRATION under U.S. GOVERNMENT Contract W-7405 ong 26 This informal document contains information which is pre- liminary and may be fragmentary or of limited scope. The assumptions, views, and conclusions expressed in this docu- ment are those of the author and are not to be interpreted as those of Union Carbide Corporation, Nuclear Division, or USERDA f IS UNLIMITED — owl Isoa- os/m sos STUDY OF MINED STORAGE CAVERNS prepared for QH10H CARBIDE MPORftTIOK Uak Ridge, Tennessee fhia icpoft jtxt fif;»!fvl afl aumini l w k ffxinurffd by ifif Umled Slaiei ftmiiuntni. u Ntith1M o Iht Uniled nor the UniffU Mjim I ner^y Rtttiith md Dtvlupmrnt A.limnuiNii,>n nor anf o( thfu e'npki)fe«t, no» an> of ttifir confraitor*k . »ul«t>ntuttu», fvf thcu employ «et, tnake» Jti» wittiMy, e»pr*<» or implied, or juumei any lejul toabilrty <» leipun^fljtj rorlhf JciUfai.),»<,fnplftcnc« or u*rulnc» of my tnfaimatiur. arParam, prudjil »r pmcrn duJuird. or tepteKnti thai U» w>utd not mCnnp pnvjttly t>»ncd JAMES H. GOBBS ENGINEERING Tulsa, Oklahoma j 1 Kl'JU i! 'T 13 UNLIMITED STUDY OF MINED STORAGE CAVERNS H. -
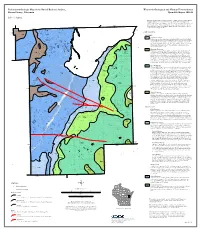
Preliminary Geologic Map of the Buried Bedrock Surface, Wisconsin Geological and Natural History Survey Brown County, Wisconsin Open-File Report 2011-02
Preliminary Geologic Map of the Buried Bedrock Surface, Wisconsin Geological and Natural History Survey Brown County, Wisconsin Open-File Report 2011-02 John A. Luczaj The bedrock surface in Brown County is typically overlain by Quaternary glacial sediment, ranging in thickness from 0 to 300 feet. These deposits include Pleistocene glacial till, outwash, and glaciolacustrine sediments from tens of feet to at least 300 feet in thickness in preglacial river valleys. Bedrock is best exposed along the Niagara Escarpment, which is 87°50'W DOOR CO 88°0'W produced by the resistant eastward-dipping Silurian Mayville Formation that overlies the ! ! ! ! BOUNDARY ! OCONTO CO!! !! ! INDEFINITE weaker Ordovician Maquoketa Shale. ! ! ! ! ! ! ! ! !! ! ! ! ! ! ! ! ! ! 88°10'W !!! ! ! ! !! ! ! ! ! ! ! ! ! L ! ! ! ! ! !! ! !! !!!! ! ! !! ! ! ! i ! ! ! ! ! ! ! !! ! t ! ! ! !! ! ! t ! ! ! ! ! ! ! ! ! 44°40'N !! ! !! ! ! !!! l ! ! ! ! ! ! ! ! e ! ! ! !!! !! ! ! ! ! ! ! ! !! ! ! ! ! ! ! ! ! ! ! T ! ! !! ! !! ! ! ! ! ! ! !! !! ! a ! ! ! ! ! ! ! ! !! ! ! ! i ! ! ! ! ! ! ! ! l ! ! ! ! ! !! P ! ! ! ! ! ! ! ! ! ! ! ! ! !!!! ! ! ! !! ! o ! ! ! ! ! ! ! ! ! ! EXPLANATION ! ! i 44°40'N ! ! ! ! ! n ! ! ! ! !!! ! ! ! ! ! !! ! ! t ! !! ! ! !! !! ! ! ! ! ! ! !! ! ! ! !! ! ! ! ! ! !! ! ! ! !!!!! ! !! ! ! ! ! ! ! ! ! Op ! ! ! ! ! ! ! ! ! ! !! ! ! ! !! ! ! ! ! ! ! ! ! ! SILURIAN ! ! ! ! ! ! ! ! ! ! ! !! ! ! ! !! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! !! ! ! ! ! ! ! ! ! ! ! Dyckesville!! Engadine Formation ! ! ! ! ! -

Index to the Geologic Names of North America
Index to the Geologic Names of North America GEOLOGICAL SURVEY BULLETIN 1056-B Index to the Geologic Names of North America By DRUID WILSON, GRACE C. KEROHER, and BLANCHE E. HANSEN GEOLOGIC NAMES OF NORTH AMERICA GEOLOGICAL SURVEY BULLETIN 10S6-B Geologic names arranged by age and by area containing type locality. Includes names in Greenland, the West Indies, the Pacific Island possessions of the United States, and the Trust Territory of the Pacific Islands UNITED STATES GOVERNMENT PRINTING OFFICE, WASHINGTON : 1959 UNITED STATES DEPARTMENT OF THE INTERIOR FRED A. SEATON, Secretary GEOLOGICAL SURVEY Thomas B. Nolan, Director For sale by the Superintendent of Documents, U.S. Government Printing Office Washington 25, D.G. - Price 60 cents (paper cover) CONTENTS Page Major stratigraphic and time divisions in use by the U.S. Geological Survey._ iv Introduction______________________________________ 407 Acknowledgments. _--__ _______ _________________________________ 410 Bibliography________________________________________________ 410 Symbols___________________________________ 413 Geologic time and time-stratigraphic (time-rock) units________________ 415 Time terms of nongeographic origin_______________________-______ 415 Cenozoic_________________________________________________ 415 Pleistocene (glacial)______________________________________ 415 Cenozoic (marine)_______________________________________ 418 Eastern North America_______________________________ 418 Western North America__-__-_____----------__-----____ 419 Cenozoic (continental)___________________________________ -

Bedrock Geology
Pbg Bedrock Geology Scale: 1:2,750,000 Pko Wisconsin Transverse Mercator NAD83(91) Map S13 - ams Pkg Pkl Avo Agn Agr Pvn Pif Pms PAgn Pku Pmg Pgn Pqz Pvu Prg Pdi Pwu Pws Pwa Opc Pwg Pgr Pwr Pwq Map Unit Names Om Cu Agn Agr W Avo Os PAgn Opc Pgn Su Pif Pdi; Pmg Oa Pvn Pqz Pvu Om Pms Du Pgr Prg Pqz Pwa Pwq Oa Pwr Pwg; Pws; Pwu Pkg Pkl Pku This information represents a digital version of the polygon units shown on "Bedrock Geologic Map of Wisconsin," originally published in 1982 at a scale of 1:1,000,000. The bedrock Pko geology shown is a lithostratigraphic interpretation of the consolidated (rock) units Pbg present at the land surface or, in most areas, the first consolidated (rock) unit encountered Cu beneath variable thicknesses of unconsolidated glacial sediment. The data include not only Opc the distribution of the various bedrock units, but also a general description of the lithologic Oa character and nomenclatural identification. Os The general descriptions of the map units appear as a table on the back of this map. Om Su Du 0 12.5 25 50 75 100 Water Miles Ecological Landscape Kilometers County Boundaries 0 20 40 80 120 160 ® Ecological Landscapes of Wisconsin Handbook - 1805.1 © WDNR, 2011 Precambrian rocks of Archean age, Upper Archean System Agn Metamorphic rocks: quartzofeldspathic gneiss, migmatite and amphibolite Agr Igneous rocks: granite and associated rocks; includes Puritan Quartz Monzonite Metamorphic rocks: mafic to intermediate metavolcanic rocks and associated metasedimentary rocks; includes Ramsey Formation in Iron -
PDF Linkchapter
Index [Italic page numbers indicate major references] Abbott Formation, Illinois, 251 Michigan, 287 beetle borrows, Nebraska, 11 Acadian belt, 429 Archean rocks beetles, Manitoba, 45 Acadian orogeny Michigan, 273, 275 Belfast Member, Brassfield Illinois, 243 Minnesota, 47, 49, 53 Formation, Ohio, 420, 421 Indiana, 359 Wisconsin, 189 Bellepoint Member, Columbus Acrophyllum oneidaense, 287 Arikaree Group, Nebraska, 13, 14, Limestone, Ohio, 396 Adams County, Ohio, 420, 431 15, 25, 28 Belleview Valley, Missouri, 160 Adams County, Wisconsin, 183 Arikareean age, Nebraska, 3 Bellevue Limestone, Indiana, 366, Admire Group, Nebraska, 37 arthropods 367 Aglaspis, 83 Iowa, 83 Bennett Member, Red Eagle Ainsworth Table, Nebraska, 5 Missouri, 137 Formation, Nebraska, 37 Alexander County, Illinois, 247, 257 Ash Hollow Creek, Nebraska, 31 Benton County, Indiana, 344 algae Ash Hollow Formation, Nebraska, 1, Benzie County, Michigan, 303 Indian, 333 2, 5, 26, 29 Berea Sandstone, Ohio, 404, 405, Michigan, 282 Ash Hollow State Historical Park, 406, 427, 428 Missouri, 137 Nebraska, 29 Berne Conglomerate, Logan Ohio, 428 asphalt, Illinois, 211 Formation, Ohio, 411, 412, 413 Alger County, Michigan, 277 Asphalting, 246 Bethany Falls Limestone Member, Algonquin age, Michigan, 286, 287 Asterobillingsa, 114 Swope Formation, Missouri, Allamakee County, Iowa, 81, 83, 84 Astrohippus, 26 135, 138 Allegheny Group, Ohio, 407 Atherton Formation, Indiana, 352 Bethany Falls Limestone, Iowa, 123 Allen County, Indiana, 328, 329, 330 athyrids, Iowa, 111 Betula, 401 Allensville -

List R - Rock Units - Alphabetical List
LIST R - ROCK UNITS - ALPHABETICAL LIST Aberystwyth Grits Ash Hollow Formation Abo Formation Ashe Formation Absaroka Supergroup Asmari Formation Acatlan Complex Astoria Formation Ackley Granite Asu River Group Acoite Formation Athabasca Formation Acungui Group Athgarh Sandstone Adamantina Formation Atoka Formation Adirondack Anorthosite Austin Chalk Admire Group** Austin Group Agbada Formation** Aux Vases Sandstone Ager Formation Avon Park Formation Agrio Formation Aycross Formation Aguacate Group Aztec Sandstone Aguja Formation Baca Formation Akiyoshi Limestone Badami Series Al Khlata Formation Bagh Beds Albert Formation Bahariya Formation Aldridge Formation Bainbridge Formation Alisitos Formation Bajo Barreal Formation Allegheny Group Baker Coal* Allen Formation* Baker Lake Group Almond Formation Bakhtiari Formation Alpine Schist* Bakken Formation** Altyn Limestone Balaklala Rhyolite Alum Shale Formation* Baldonnel Formation Ambo Group** Ballachulish Complex* Ameki Formation Ballantrae Complex Americus Limestone Member Baltimore Gneiss Ames Limestone Bambui Group Amisk Group Banded Gneissic Complex Amitsoq Gneiss Bandelier Tuff Ammonoosuc Volcanics Banff Formation Amsden Formation Bangor Limestone Anahuac Formation Banquereau Formation* Andalhuala Formation Banxi Group Andrew Formation* Baota Formation Animikie Group Baquero Formation Annot Sandstone Barabash Suite Anshan Group Baraboo Quartzite Antalya Complex Baraga Group Antelope Shale Barail Group Antelope Valley Limestone Baralaba Coal Measures Antietam Formation Barnett Shale -

An Evaluation of the Shallow Ground-Water Resources in Will and Southern Cook Counties, Illinois
ISWS RR 123 copy 1 loan Report 123 Meeting the Growing Demand for Water: An Evaluation of the Shallow Ground-Water Resources in Will and Southern Cook Counties, Illinois by George S. Roadcap, Stuart J. Cravens, and Edward C. Smith ILLINOIS STATE WATER SURVEY DEPARTMENT OF ENERGY AND NATURAL RESOURCES 1993 DATE DUE 5/28/97 10 06 98 4/2/2000 11/17/00 ILLlNOIS STATE WATER SURVEY LIBRARY 2204 GRIFFITH DRIVE CHAMPAIGN, 1L 61820 ISWS RR Roadcap, George S. 123 MEETING THE GROWING copy 1 DEMAND FOR WATER: AN loan EVALUATION OF THE aaa4735 SHALLOW GROUND-WATER RESOURCES IN WILL AND DEMCO RESEARCH REPORT 123 Meeting the Growing Demand for Water: An Evaluation of the Shallow Ground-Water Resources in Will and Southern Cook Counties, Illinois George S. Roadcap, Stuart J. Cravens, and Edward C. Smith Title: Meeting the Growing Demand for Water: An Evaluation of the Shallow Ground-Water Resources in Will and Southern Cook Counties, Illinois. Abstract: This study evaluates the heavily used shallow aquifer system in Will and southern Cook Counties and ascertains its ability to meet the present and future water supply needs of the communities it serves. The hydrogeologic evaluation involves an examination of existing data to determine the aquifer's geologic and hydraulic properties as well as the amount of water withdrawn from the aquifer. New data are used to determine ground-water flow directions, existing aquifer recharge rates, potential yield of the aquifer, and general ground-water quality. Reference: Roadcap G.S., S.J. Cravens, and E.C. Smith. Meeting the Growing Demand for Water: An Evaluation of the Shallow Ground-Water Resources in Will and Southern Cook Counties, Illinois.