Evaluation of B Lymphocyte Deficiencies
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-
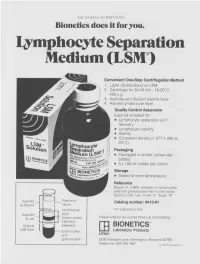
Lymphocyte Separation Medium (LSM
THE JOURNAL OF IMMUNOLOGY Bionetics does it for you. Lymphocyte Separation Medium (LSM wenient One-Step Centrifugation Method _ayer diluted blood on LSM. 2,entrifuge for 30-40 min., 18-20°C, ~.00 x g. ~,spirate and discard plasma layer -larvest lymphocyte layer. Quality Control Assurance Each lot is tested for: • Lymphocyte separation and recovery. • Lymphocyte viability. • Sterility. • Consistent density (1.077-1.080 at 20°C). Packaging • Packaged in amber, screw-cap bottles. • 5 x 100 ml bottles per carton. Storage. • Stored at room temperature. Reference Boyum, A. (1968): Isolation of mononuclear cells and granulocytes from human blood. Scand J. Clin. Lab. Invest. 21, Suppl. 97. Aspirate I IC[OI I IC~ LJI Catalog number: 8410-01 & discard serum Lymphocyte For Laboratory Use Aspirate layer & use (mononuclear Please write for our current Price List and Catalog. cells and Original platelets) ITi BIONETICS° LSM layer Erythrocytes Laboratory Products and Litton granulocytes 5516 Nicholson Lane, Kensington, Maryland 20795 Telephone: (301) 881-1557 1979 Litton Bionetics, tnc Get the most out of your high quality cytotoxic antibodies with LOW-TOX-M RABBIT COMPLEMENT LOW TOXICITY HIGH ACTIVITY Presentation: CL 3051 5 x 1 ml, lyophilized $30.00 When it comes to COMPLEMENT... come to CEDARLANE Direct orders or inquiries to: UNITED STATES: WORLDWIDE EXCEPT U.S. ,4 C~L CEDARLANE ACCURATE CHEMICAL & LABORATO RI ES SCIENTIFIC CORPORATION LIMITED 5516-8TH LINE, R.R. 2 28 TEC STREET, HICKSVtLLE, N.Y. 11801 HORNBY, ONTARIO, CANADA LOP 1E0 Telephone -

Stress and the Immune System Tracy B
4 World Health • 47th Yeor, No. 2, Morch-Aprill994 Stress and the immune system Tracy B. Herbert any people have the effects of factors as diverse as experienced the examinations, bereavement, divorce, Mconnection between stress unemployment, mental arithmetic, and getting sick. Colds, influenza, and looking after a relative with herpes and allergies seem worse Alzheimer's di sease. In general, when we are severely stressed at these studies find that stress is work or in the home. Others are related to changes in both the never sick until they go on vacation numbers of white blood cells in (that is, after the stress is over), and circulation and the quantity of then they spend the whole time antibody in the blood. Moreover, fighting the virus. Because of stress is associated with changes in intrinsic connections like these, the functioning of immune cells. many researchers are today That is, there is a relatively large exploring whether (and how) stress decrease in both lymphocyte and illness are actually linked. One proliferation and natural killer cell specific focus of this research is to activity in individuals who have study the effects of stress on the experienced stress. There seems to immune systems; after all, if stress A lymphocyte: stress may weaken the capacity be some connection between the affects immunity, that would be one of lymphocytes to combat infection. duration of the stress and the amount way in which stress could contribute of immune change. For example, the to illness. longer the stress, the greater the The function of the immune proliferation"- by incubating these decrease in the number of specific system is to protect us from cells for several days with types of white blood cells. -

Association Between Neutrophil-Lymphocyte Ratio and Herpes Zoster Infection in 1688 Living Donor Liver Transplantation Recipients at a Large Single Center
biomedicines Article Association between Neutrophil-Lymphocyte Ratio and Herpes Zoster Infection in 1688 Living Donor Liver Transplantation Recipients at a Large Single Center Ji-Hoon Sim, Young-Jin Moon, Sung-Hoon Kim, Kyoung-Sun Kim , Ju-Seung Lee, Jun-Gol Song * and Gyu-Sam Hwang Department of Anesthesiology and Pain Medicine, Asan Medical Center, University of Ulsan College of Medicine, Seoul 05505, Korea; [email protected] (J.-H.S.); [email protected] (Y.-J.M.); [email protected] (S.-H.K.); [email protected] (K.-S.K.); [email protected] (J.-S.L.); [email protected] (G.-S.H.) * Correspondence: [email protected]; Tel.: +82-2-3010-3869 Abstract: Liver transplantation (LT) is closely associated with decreased immune function, a contrib- utor to herpes zoster (HZ). However, risk factors for HZ in living donor LT (LDLT) remain unknown. Neutrophil-lymphocyte ratio (NLR) and immune system function are reportedly correlated. This study investigated the association between NLR and HZ in 1688 patients who underwent LDLT between January 2010 and July 2020 and evaluated risk factors for HZ and postherpetic neuralgia (PHN). The predictive power of NLR was assessed through the concordance index and an integrated discrimination improvement (IDI) analysis. Of the total cohort, 138 (8.2%) had HZ. The incidence of HZ after LT was 11.2 per 1000 person-years and 0.1%, 1.3%, 2.9%, and 13.5% at 1, 3, 5, and 10 years, Citation: Sim, J.-H.; Moon, Y.-J.; Kim, respectively. In the Cox regression analysis, preoperative NLR was significantly associated with HZ S.-H.; Kim, K.-S.; Lee, J.-S.; Song, J.-G.; (adjusted hazard ratio [HR], 1.05; 95% confidence interval [CI], 1.02–1.09; p = 0.005) and PHN (HR, Hwang, G.-S. -

Lymphocyte-Activation Gene 3 (LAG-3) Immune Pathway
Lymphocyte-Activation Gene 3 (LAG-3) About LAG-3 LAG-3 Lymphocyte-activation gene 3 (LAG-3) is an immune checkpoint receptor protein found on the cell surface of effector T cells and regulatory T cells (Tregs) and functions to control T cell response, activation and growth.1 TCR T cells are a type of white blood cell that are part of the immune system. Activation of cytotoxic T cells by antigens enables them to 1 kill unhealthy or foreign cells. Inactive T cell Antigen MHC Dendritic cell (APC) LAG-3 and LAG-3 and LAG-3 and Immune Function T Cell Exhaustion Cancer • After a T cell is activated to kill its • However, in certain situations where T • Because of its critical role in regulating target cell, LAG-3 expression is cells experience prolonged exposure to an exhaustion of cytotoxic T cells and Treg increased to turn off the immune antigen, such as cancer or chronic function, LAG-3 has become a target of response, so that the T cell does not go infection, the T cells become desensitized study in the cancer field. on to attack healthy cells.2 and lose their ability to activate and multiply in the presence of the antigen.4 • In cancer, LAG-3 expressing exhausted • Inhibition of the immune response is cytotoxic T cells and Tregs expressing accomplished through activation of • The desensitized T cell will also LAG-3 gather at tumor sites.5,6 the LAG-3 pathway, which can occur progressively fail to produce cytokines via binding of LAG-3 to a type of (proteins that assist in the immune • Preclinical studies suggest that inhibiting antigen-presenting complex called response) and kill the target cells.4 LAG-3 allows T cells to regain their MHC II. -

Understanding the Immune System: How It Works
Understanding the Immune System How It Works U.S. DEPARTMENT OF HEALTH AND HUMAN SERVICES NATIONAL INSTITUTES OF HEALTH National Institute of Allergy and Infectious Diseases National Cancer Institute Understanding the Immune System How It Works U.S. DEPARTMENT OF HEALTH AND HUMAN SERVICES NATIONAL INSTITUTES OF HEALTH National Institute of Allergy and Infectious Diseases National Cancer Institute NIH Publication No. 03-5423 September 2003 www.niaid.nih.gov www.nci.nih.gov Contents 1 Introduction 2 Self and Nonself 3 The Structure of the Immune System 7 Immune Cells and Their Products 19 Mounting an Immune Response 24 Immunity: Natural and Acquired 28 Disorders of the Immune System 34 Immunology and Transplants 36 Immunity and Cancer 39 The Immune System and the Nervous System 40 Frontiers in Immunology 45 Summary 47 Glossary Introduction he immune system is a network of Tcells, tissues*, and organs that work together to defend the body against attacks by “foreign” invaders. These are primarily microbes (germs)—tiny, infection-causing Bacteria: organisms such as bacteria, viruses, streptococci parasites, and fungi. Because the human body provides an ideal environment for many microbes, they try to break in. It is the immune system’s job to keep them out or, failing that, to seek out and destroy them. Virus: When the immune system hits the wrong herpes virus target or is crippled, however, it can unleash a torrent of diseases, including allergy, arthritis, or AIDS. The immune system is amazingly complex. It can recognize and remember millions of Parasite: different enemies, and it can produce schistosome secretions and cells to match up with and wipe out each one of them. -

Early Lymphocyte Recovery Predicts Longer Survival After Autologous Peripheral Blood Stem Cell Transplantation in Multiple Myeloma
Bone Marrow Transplantation (2006) 37, 1037–1042 & 2006 Nature Publishing Group All rights reserved 0268-3369/06 $30.00 www.nature.com/bmt ORIGINAL ARTICLE Early lymphocyte recovery predicts longer survival after autologous peripheral blood stem cell transplantation in multiple myeloma H Kim1, H-J Sohn2, S Kim2, J-S Lee2, W-K Kim2 and C Suh2 1Division of Hematology-Oncology, Ulsan University Hospital, University of Ulsan College of Medicine, Ulsan, Korea and 2Division of Oncology, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea To understand the prognostic value of lymphocyte Although the use of allogeneic hematopoietic stem cell recovery after autologous peripheral blood stem cell transplantation has been increasing, autologous stem cell transplantation (APBSCT), we performed a retrospective transplantation (ASCT) remains the standard therapeutic study of 59 newly diagnosed multiple myeloma (MM) modality for MM.1–3 The main drawback of ASCT is its patients who underwent frontline APBSCT. Conditioning high relapse rate. Because immune-mediated tumor eradi- regimens were melphalan 100 mg/m2 for 2 days. Follow- cation may lower the relapse rate associated with ASCT, ing APBSCT, all patients showed complete or partial there is increasing interest in immune reconstitution response. Median follow-up time was 29.57 months and following ASCT.4–9 In support of this, a relationship median recovery of absolute lymphocyte count (ALC) between lymphocyte recovery after ASCT and relapse rate X1000/mm3 was 23 days. Univariate analysis revealed or survival has been observed in many diseases, suggesting that significant predictors of overall survival (OS) that early lymphocyte recovery associated with immune included bone marrow (BM) plasma cells p40% at reconstitution can act against residual disease progres- diagnosis (P ¼ 0.0243) and recovery of ALC X1000/mm3 sion.10–17 by day þ 23 (P ¼ 0.0156). -

The CLL Guide Information for Patients and Caregivers Chronic Lymphocytic Leukemia
The CLL Guide Information for Patients and Caregivers Chronic Lymphocytic Leukemia Laura, CLL survivor This publication was supported by Revised 2014 Inside Front Cover A Message from Louis J. DeGennaro, PhD President and CEO of The Leukemia & Lymphoma Society The Leukemia & Lymphoma Society (LLS) is the world’s largest voluntary health organization dedicated to finding cures for blood cancer patients. Since 1954, we have invested over $1 billion in research specifically targeting blood cancers to advance therapies and save lives. We will continue to invest in research for cures, programs and services to improve the quality of life for people with chronic lymphocytic leukemia (CLL). We know that understanding CLL can be tough. We are here to help and are committed to provide you with the most up-to-date information about CLL, your treatment and your support options. We know how important it is for you to understand your health information and to use it with your healthcare team toward good health, remission and recovery. Our vision is that one day all people with CLL will be cured or be able to manage their disease with good quality of life. Until then, we trust the information in this Guide will help you along your journey. We wish you well. Louis J. DeGennaro, PhD President and Chief Executive Officer The Leukemia & Lymphoma Society Inside This Guide 2 Introduction 3 Resources and Information 7 Part 1—Understanding CLL About Blood The Immune System What is CLL? Signs and Symptoms Diagnosing CLL Tracking Your CLL Tests 14 Part 2—Treating CLL Finding the Right Doctor Treatment Planning Treatments for CLL Treatment for Relapsed or Refractory CLL Stem Cell Transplantation 26 Part 3—About Clinical Trials 27 Part 4—Side Effects and Treatment Response Side Effects of CLL Treatment Treatment Response 29 Follow-up Care 30 Ongoing Care 31 Health Terms 34 Healthcare Question Guides This LLS guide about CLL is for information only. -

B-Cell Development, Activation, and Differentiation
B-Cell Development, Activation, and Differentiation Sarah Holstein, MD, PhD Nov 13, 2014 Lymphoid tissues • Primary – Bone marrow – Thymus • Secondary – Lymph nodes – Spleen – Tonsils – Lymphoid tissue within GI and respiratory tracts Overview of B cell development • B cells are generated in the bone marrow • Takes 1-2 weeks to develop from hematopoietic stem cells to mature B cells • Sequence of expression of cell surface receptor and adhesion molecules which allows for differentiation of B cells, proliferation at various stages, and movement within the bone marrow microenvironment • Immature B cell leaves the bone marrow and undergoes further differentiation • Immune system must create a repertoire of receptors capable of recognizing a large array of antigens while at the same time eliminating self-reactive B cells Overview of B cell development • Early B cell development constitutes the steps that lead to B cell commitment and expression of surface immunoglobulin, production of mature B cells • Mature B cells leave the bone marrow and migrate to secondary lymphoid tissues • B cells then interact with exogenous antigen and/or T helper cells = antigen- dependent phase Overview of B cells Hematopoiesis • Hematopoietic stem cells (HSCs) source of all blood cells • Blood-forming cells first found in the yolk sac (primarily primitive rbc production) • HSCs arise in distal aorta ~3-4 weeks • HSCs migrate to the liver (primary site of hematopoiesis after 6 wks gestation) • Bone marrow hematopoiesis starts ~5 months of gestation Role of bone -

Editorial Overview: Lymphocyte Development and Activation: Lymphocytes, Integrators of Information
COIMMU-1429; NO. OF PAGES 3 Available online at www.sciencedirect.com ScienceDirect Editorial overview: Lymphocyte development and activation: Lymphocytes, integrators of information Jeroen Roose and Sidonia Fagarasan Current Opinion in Immunology 2015, 33:xx–yy http://dx.doi.org/10.1016/j.coi.2015.03.003 0952-7915/# 2015 Elsevier Ltd. All rights reserved. Jeroen Roose Research on lymphocyte development and activation has matured to a level of sophistication that we can start to integrate multiple instructions coming from various players and understand how these instructive ‘networks’ govern lymphocyte function. The collection of reviews in this edition of Current Opinion in Immunology focuses on the complexity of intracellular cues that overcome a signal threshold paired with changes in metabolic programs. In the second section recent advances on cell-extrinsic factors and cell–cell interactions as regulators of lymphocyte function are highlighted. Department of Anatomy, University of California, San Francisco (UCSF), CA, USA Stimulation of antigen receptors on lymphocytes leads to triggering of e-mail: [email protected] intracellular signaling pathways that are connected to form signaling net- Jeroen Roose is an associate professor at works. The topology and dynamic nature of these networks are only just UCSF. His laboratory has contributed to the emerging. Helou and Salomon provide an overview of the current state of concept that lymphocytes can transduce high-resolution mass spectrometry and how it has enabled to understand analog or digital Ras-MAP kinase signals these networks in a quantitative and qualitative manner. Their phospho- depending on the strength of receptor signal proteomic approaches on T cells have provided us with a blueprint of the and the choice of Ras activator. -

Lymphocyte Recirculation
Lymphocyte Recirculation Chapter 15 Naïve lymphocytes enter lymph nodes Lymphocyte Migration and from the blood circulation Lymphocytes return Inflammation to blood via the thoracic duct Antigens from infected area go to lymph nodes via the lymphatic system I. Primary Lymphoid II. Secondary - Lymph Nodes -Spleen -MALT Lymphatic vessels Leukocytes are constantly moving between sites where - Collect interstitial fluid and carry antigens may be It (lymph), via a system of encountered: progressively larger vessels, into regional lymph nodes. 1 2 - Spleen (via afferent lymphatic vessels). - Lymph nodes - Other secondary -Lymph leaves the lymph nodes lymphoid tissues via efferent lymphatic vessels, - Other tissues – which eventually drain back especially skin and into the circulatory system (via mucosal surfaces the thoracic duct). 3 Leukocytes accumulate at sites of inflammation 1 CHOICES: Antigen capture: (APC) or 1) If no antigen is present: lymphocytes Macrophages: capture and process particulate routinely enter and leave secondary lymphoid antigens (via phagocytosis) tissues Dendritic cells: capture and process non- 2) If antigen enters the secondary lymphoid particulate antigens (via endocytosis) tissue: Lymphocyte proliferation in response to antigen B cells: capture and process antigens that bind to occurs within the lymphoid tissue. surface BCR (via endocytosis) After several days, antigen-activated lymphocytes begin leaving the lymphoid tissue. Dendritic cells: originate in bone marrow, capture antigen within tissues and transport antigen to secondary lymphoid tissue. Lymphocytes can enter lymphoid tissues in two ways: 1 1) Direct entry into lymph nodes via afferent lymphatics 2) Entry from blood capillaries across specialized endothelial cells (high-walled endothelial cells) present in the postcapillary venules (High Endothelial Venules= HEV) within the secondary 2 lymphoid tissue. -

Lymphocyte Trapping
Immunology 1976 30 749 Lymphocyte trapping DIFFERENTIAL EFFECTS OF ATS AND IRRADIATION ON TRAPPING IN LYMPH NODES AND SPLEEN MARION M. ZATZ Departments of Microbiology and Surgery, Yale University School of Medicine, New Haven, Connecticut, U.S.A. Received 20 August 1975; acceptedfor publication 13 November 1975 Summary. Increased sequestration (trapping) of lymphocytes in lymphoid organs (Zatz and Lance, lymphocytes occurs in lymphoid organs following 1971; Ford, 1972; Hall and Morris, 1965; Rowley, antigenic challenge. The effects of irradiation and Gowans, Atkins, Ford and Smith, 1972). This anti-thymocyte serum (ATS) upon lymphocyte sequestration of cells has been termed lymphocyte trapping in lymph nodes and spleen were studied. 'trapping' (Zatz and Lance, 1971). Both the above agents diminished or abrogated It seems likely that lymphocyte trapping indirectly trapping in the draining lymph nodes; these same plays an important role in vivo in the regulation of a agents resulted in enhanced trapping in the spleen. broad spectrum of immune responses, via control of Suppression of lymph node trapping could be lymphocyte traffic (Zatz and Lance, 1971; Ford, achieved with a low dose of ATS and with 850 R, 1972; Hall and Morris, 1965; Rowley et al., 1972; but not 200 R. Emeson and Thursh, 1971; Dresser, Taub and These results are interpreted as showing that Krantz, 1970; Zatz and Lance, 1971; Zatz, White lymphocyte trapping in lymph nodes is initiated and Goldstein, 1973; Gershon and Fightlin, 1973; primarily by an ATS- and irradiation-sensitive Gillette and Bellanti, 1973; Asherson and Barnes, population, whereas in the spleen a cell population 1973; Frost and Lance, 1973; Zatz and Gershon, resistant to the above agents activates the lymphocyte 1974). -

I M M U N O L O G Y Core Notes
II MM MM UU NN OO LL OO GG YY CCOORREE NNOOTTEESS MEDICAL IMMUNOLOGY 544 FALL 2011 Dr. George A. Gutman SCHOOL OF MEDICINE UNIVERSITY OF CALIFORNIA, IRVINE (Copyright) 2011 Regents of the University of California TABLE OF CONTENTS CHAPTER 1 INTRODUCTION...................................................................................... 3 CHAPTER 2 ANTIGEN/ANTIBODY INTERACTIONS ..............................................9 CHAPTER 3 ANTIBODY STRUCTURE I..................................................................17 CHAPTER 4 ANTIBODY STRUCTURE II.................................................................23 CHAPTER 5 COMPLEMENT...................................................................................... 33 CHAPTER 6 ANTIBODY GENETICS, ISOTYPES, ALLOTYPES, IDIOTYPES.....45 CHAPTER 7 CELLULAR BASIS OF ANTIBODY DIVERSITY: CLONAL SELECTION..................................................................53 CHAPTER 8 GENETIC BASIS OF ANTIBODY DIVERSITY...................................61 CHAPTER 9 IMMUNOGLOBULIN BIOSYNTHESIS ...............................................69 CHAPTER 10 BLOOD GROUPS: ABO AND Rh .........................................................77 CHAPTER 11 CELL-MEDIATED IMMUNITY AND MHC ........................................83 CHAPTER 12 CELL INTERACTIONS IN CELL MEDIATED IMMUNITY ..............91 CHAPTER 13 T-CELL/B-CELL COOPERATION IN HUMORAL IMMUNITY......105 CHAPTER 14 CELL SURFACE MARKERS OF T-CELLS, B-CELLS AND MACROPHAGES...............................................................111