Mutated SF3B1 Is Associated with Transcript Isoform Changes of The
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Anti-Phospho-SF3B1 (Sap155) (Ser129) Pab
PD043 Page 1 For Research Use Only. Not for use in diagnostic procedures. Anti-Phospho-SF3B1 (Sap155) (Ser129) pAb CODE No. PD043 CLONALITY Polyclonal ISOTYPE Rabbit Ig, affinity purified QUANTITY 100 L SOURCE Purified IgG from rabbit serum IMMUNOGEN KLH conjugated synthetic peptide, RTMII(pS)PERL (corresponding to amino acid residues 124-133 of human SF3B1) FORMURATION PBS containing 50% Glycerol (pH 7.2). No preservative is contained. STORAGE This antibody solution is stable for one year from the date of purchase when stored at -20°C. APPLICATION-CONFIRMED Western blotting 1:1000 for chemiluminescence detection system SPECIES CROSS REACTIVITY on WB Species Human Mouse Rat Hamster Cell HeLa, Raji NIH/3T3, WR19L Not tested Not tested Reactivity Entrez Gene ID 23451 (Human), 81898 (Mouse) REFERENCES 1) Wang, C., et al., Genes Dev. 12, 1409-1414 (1998) 2) Shi, Y., et al., Mol Cell 23, 819-829 (2006) 3) Tanuma, N., et al., J. Biol. Chem., 283, 35805-35814 (2008) 4) Yoshida, K., et al., Nature 478, 64-69 (2011) 5) Rossi, D., et al., Blood 118, 6904-6908 (2011) 6) Quesada, V., et al., Nat Genet 44, 47-52 (2011) 7) Wang, L., et al., N Engl J Med. 365, 2497-2506 (2011) 8) Biankin, A. V., et al., Nature 491, 399-405 (2012) For more information, please visit our web site http://ruo.mbl.co.jp/ MEDICAL & BIOLOGICAL LABORATORIES CO., LTD. URL http://ruo.mbl.co.jp/ e-mail [email protected], TEL 052-238-1904 PD043 Page 2 P a g RELATED PRODUCTS e PD043 Anti-Phospho-SF3B1 (Sap155) (Ser129) pAb D138-3 Anti-Sap155 (SF3B12) mAb D221-3 Anti-Sap155 (SF3B1) -

Monoclonal B-Cell Lymphocytosis Is Characterized by Mutations in CLL Putative Driver Genes and Clonal Heterogeneity Many Years Before Disease Progression
Leukemia (2014) 28, 2395–2424 © 2014 Macmillan Publishers Limited All rights reserved 0887-6924/14 www.nature.com/leu LETTERS TO THE EDITOR Monoclonal B-cell lymphocytosis is characterized by mutations in CLL putative driver genes and clonal heterogeneity many years before disease progression Leukemia (2014) 28, 2395–2398; doi:10.1038/leu.2014.226 (Beckton Dickinson) and data analyzed using Cell Quest software. On the basis of FACS (fluorescence-activated cell sorting) analysis, we observed after enrichment an average of 91% of CD19+ cells Monoclonal B-cell lymphocytosis (MBL) is defined as an asympto- (range 76–99%) and 91% of the CD19+ fraction were CD19+/CD5+ matic expansion of clonal B cells with less than 5 × 109/L cells in the cells (range 66–99%). We used the values of the CD19+/CD5+ peripheral blood and without other manifestations of chronic fraction to calculate the leukemic B-cell fraction and reduce any lymphocytic leukemia (CLL; for example, lymphadenopathy, cyto- significant contamination of non-clonal B cells in each biopsy. DNA penias, constitutional symptoms).1 Approximately 1% of the MBL was extracted from the clonal B cells and non-clonal (that is, T cells) cohort develops CLL per year. Evidence suggests that nearly all CLL cells using the Gentra Puregene Cell Kit (Qiagen, Hilden, Germany). 2 fi fi cases are preceded by an MBL state. Our understanding of the Extracted DNAs were ngerprinted to con rm the relationship genetic basis, clonal architecture and evolution in CLL pathogenesis between samples of the same MBL individual and to rule out sample has undergone significant improvements in the last few years.3–8 In cross-contamination between individuals. -

SF3B2-Mediated RNA Splicing Drives Human Prostate Cancer Progression
Published OnlineFirst August 20, 2019; DOI: 10.1158/0008-5472.CAN-18-3965 Cancer Molecular Cell Biology Research SF3B2-Mediated RNA Splicing Drives Human Prostate Cancer Progression Norihiko Kawamura1,2, Keisuke Nimura1, Kotaro Saga1, Airi Ishibashi1, Koji Kitamura1,3, Hiromichi Nagano1, Yusuke Yoshikawa4, Kyoso Ishida1,5, Norio Nonomura2, Mitsuhiro Arisawa4, Jun Luo6, and Yasufumi Kaneda1 Abstract Androgen receptor splice variant-7 (AR-V7) is a General RNA splicing SF3B2 complex-mediated alternative RNA splicing constitutively active AR variant implicated in U2 castration-resistant prostate cancers. Here, we show U2 snRNA that the RNA splicing factor SF3B2, identified by 3’ 3’ in silico and CRISPR/Cas9 analyses, is a critical 5’ 3’ splice site 5’ SF3B7 AR-V7 5’ A U2AF2 AGA Exon ? determinant of expression and is correlated SF3B6(p14) SF3B4 SF3B1 SF3B4 SF3B1 with aggressive cancer phenotypes. Transcriptome SF3B5 SF3B2 SF3B3 SF3B2 SF3B3 and PAR-CLIP analyses revealed that SF3B2 con- SF3A3 SF3B2 complex SF3A3 SF3A1 SF3A1 SF3b complex trols the splicing of target genes, including AR, to AR pre-mRNA drive aggressive phenotypes. SF3B2-mediated CE3 aggressive phenotypes in vivo were reversed by AR-V7 mRNA AR mRNA AR-V7 knockout. Pladienolide B, an inhibitor of CE3 a splicing modulator of the SF3b complex, sup- Drive malignancy pressed the growth of tumors addicted to high While the SF3b complex is critical for general RNA splicing, SF3B2 promotes inclusion of the target exon through recognizing a specific RNA motif. SF3B2 expression. These findings support the idea © 2019 American Association for Cancer Research that alteration of the splicing pattern by high SF3B2 expression is one mechanism underlying prostate cancer progression and therapeutic resistance. -

Flexible, Unbiased Analysis of Biological Characteristics Associated with Genomic Regions
bioRxiv preprint doi: https://doi.org/10.1101/279612; this version posted March 22, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. BioFeatureFinder: Flexible, unbiased analysis of biological characteristics associated with genomic regions Felipe E. Ciamponi 1,2,3; Michael T. Lovci 2; Pedro R. S. Cruz 1,2; Katlin B. Massirer *,1,2 1. Structural Genomics Consortium - SGC, University of Campinas, SP, Brazil. 2. Center for Molecular Biology and Genetic Engineering - CBMEG, University of Campinas, Campinas, SP, Brazil. 3. Graduate program in Genetics and Molecular Biology, PGGBM, University of Campinas, Campinas, SP, Brazil. *Corresponding author: [email protected] Mailing address: Center for Molecular Biology and Genetic Engineering - CBMEG, University of Campinas, Campinas, SP, Brazil. Av Candido Rondo, 400 Cidade Universitária CEP 13083-875, Campinas, SP Phone: 55-19-98121-937 bioRxiv preprint doi: https://doi.org/10.1101/279612; this version posted March 22, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Abstract BioFeatureFinder is a novel algorithm which allows analyses of many biological genomic landmarks (including alternatively spliced exons, DNA/RNA- binding protein binding sites, and gene/transcript functional elements, nucleotide content, conservation, k-mers, secondary structure) to identify distinguishing features. -

Full-Length Transcript Characterization of SF3B1 Mutation in Chronic Lymphocytic Leukemia Reveals Downregulation of Retained Introns
ARTICLE https://doi.org/10.1038/s41467-020-15171-6 OPEN Full-length transcript characterization of SF3B1 mutation in chronic lymphocytic leukemia reveals downregulation of retained introns Alison D. Tang 1, Cameron M. Soulette 2, Marijke J. van Baren1, Kevyn Hart1, Eva Hrabeta-Robinson1, ✉ Catherine J. Wu 3,4,5 & Angela N. Brooks 1 SF3B1 1234567890():,; While splicing changes caused by somatic mutations in are known, identifying full- length isoform changes may better elucidate the functional consequences of these mutations. We report nanopore sequencing of full-length cDNA from CLL samples with and without SF3B1 mutation, as well as normal B cell samples, giving a total of 149 million pass reads. We present FLAIR (Full-Length Alternative Isoform analysis of RNA), a computational workflow to identify high-confidence transcripts, perform differential splicing event analysis, and dif- ferential isoform analysis. Using nanopore reads, we demonstrate differential 3’ splice site changes associated with SF3B1 mutation, agreeing with previous studies. We also observe a strong downregulation of intron retention events associated with SF3B1 mutation. Full-length transcript analysis links multiple alternative splicing events together and allows for better estimates of the abundance of productive versus unproductive isoforms. Our work demon- strates the potential utility of nanopore sequencing for cancer and splicing research. 1 Department of Biomolecular Engineering, University of California, Santa Cruz, CA 95062, USA. 2 Department of Molecular Cell & Developmental Biology, University of California, Santa Cruz, CA 95062, USA. 3 Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, MA 02215, USA. 4 Broad Institiute of Harvard and MIT, Cambridge, MA, USA. -
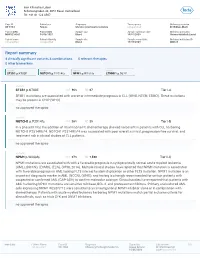
Report Summary 4 Clinically Significant Variants & Combinations 0 Relevant Therapies 0 Other Biomarkers
Inov Klinisches Labor Schützengraben 42, 4051 Basel, Switzerland Tel: +41 61 123 4567 Case ID Patient sex Diagnosis Tumor purity Ordering physician XB-1064 Female Chronic lymphocytic leukemia not specified Dr. Niklaus Marti Patient MRN Patient DOB Sample type Sample collection date Ordering institution MRN1234567 04/08/1957 Blood 10/01/2020 Universitätsklinik Liestal Patient name Patient ethnicity Sample site Sample receipt date Ordering institution ID not specified not specified Blood 10/03/2020 OID123 Report summary 4 clinically significant variants & combinations 0 relevant therapies 0 other biomarkers variant variant variant variant SF3B1 p.K700E NOTCH1 p.P2514fs NPM1 p.W288fs CTNNB1 p.D32Y variant SF3B1 p.K700E VAF 36% RD 67 Tier I-A SF3B1 mutations are associated with worse or intermediate prognosis in CLL (WHO, NCCN, ESMO). These mutations may be present in CHIP (WHO). no approved therapies variant NOTCH1 p.P2514fs VAF 28% RD 25 Tier I-B In a phase III trial, the addition of rituximab to FC chemotherapy showed no benefit in patients with CLL harboring NOTCH1 P2514Rfs*4. NOTCH1 P2514Rfs*4 was associated with poor overall survival, progression-free survival, and treatment risk in clinical studies of CLL patients. no approved therapies variant NPM1 p.W288fs VAF 27% RD 1,590 Tier II-C NPM1 mutations are associated with with a favorable prognosis in cytogenetically normal acute myeloid leukemia (AML), (NCCN), (ESMO), (ELN), (WHO, 2016). Multiple clinical studies have reported that NPM1 mutation is associated with favorable prognosis in AML lacking FLT3 internal tandem duplication or other FLT3 mutation. NPM1 mutation is an important diagnostic marker in AML (NCCN), (WHO), and testing is strongly recommended for certain patients with suspected or confirmed AML (CAP-ASH) to confirm molecular subtype. -

Cryptic Splicing Events in the Iron Transporter ABCB7 and Other Key Target Genes in SF3B1-Mutant Myelodysplastic Syndromes
OPEN Leukemia (2016) 30, 2322–2331 www.nature.com/leu ORIGINAL ARTICLE Cryptic splicing events in the iron transporter ABCB7 and other key target genes in SF3B1-mutant myelodysplastic syndromes H Dolatshad1,2,8, A Pellagatti1,2,8, FG Liberante3, M Llorian4, E Repapi5, V Steeples1,2, S Roy1,2, L Scifo1,2, RN Armstrong1,2, J Shaw1,2, BH Yip1,2, S Killick6,RKušec7, S Taylor5, KI Mills3, KI Savage3, CWJ Smith4 and J Boultwood1,2 The splicing factor SF3B1 is the most frequently mutated gene in myelodysplastic syndromes (MDS), and is strongly associated with the presence of ring sideroblasts (RS). We have performed a systematic analysis of cryptic splicing abnormalities from RNA sequencing data on hematopoietic stem cells (HSCs) of SF3B1-mutant MDS cases with RS. Aberrant splicing events in many downstream target genes were identified and cryptic 3′ splice site usage was a frequent event in SF3B1-mutant MDS. The iron transporter ABCB7 is a well-recognized candidate gene showing marked downregulation in MDS with RS. Our analysis unveiled aberrant ABCB7 splicing, due to usage of an alternative 3′ splice site in MDS patient samples, giving rise to a premature termination codon in the ABCB7 mRNA. Treatment of cultured SF3B1-mutant MDS erythroblasts and a CRISPR/Cas9-generated SF3B1-mutant cell line with the nonsense-mediated decay (NMD) inhibitor cycloheximide showed that the aberrantly spliced ABCB7 transcript is targeted by NMD. We describe cryptic splicing events in the HSCs of SF3B1-mutant MDS, and our data support a model in which NMD-induced downregulation of the iron exporter ABCB7 mRNA transcript resulting from aberrant splicing caused by mutant SF3B1 underlies the increased mitochondrial iron accumulation found in MDS patients with RS. -

Roles of Splicing Factors in Hormone-Related Cancer Progression
International Journal of Molecular Sciences Review Roles of Splicing Factors in Hormone-Related Cancer Progression Toshihiko Takeiwa 1, Yuichi Mitobe 1, Kazuhiro Ikeda 1, Kuniko Horie-Inoue 1 and Satoshi Inoue 1,2,* 1 Division of Gene Regulation and Signal Transduction, Research Center for Genomic Medicine, Saitama Medical University, Hidaka, Saitama 350-1241, Japan; [email protected] (T.T.); [email protected] (Y.M.); [email protected] (K.I.); [email protected] (K.H.-I.) 2 Department of Systems Aging Science and Medicine, Tokyo Metropolitan Institute of Gerontology, Itabashi-ku, Tokyo 173-0015, Japan * Correspondence: [email protected]; Tel.: +81-3-3964-3241 Received: 8 February 2020; Accepted: 20 February 2020; Published: 25 February 2020 Abstract: Splicing of mRNA precursor (pre-mRNA) is a mechanism to generate multiple mRNA isoforms from a single pre-mRNA, and it plays an essential role in a variety of biological phenomena and diseases such as cancers. Previous studies have demonstrated that cancer-specific splicing events are involved in various aspects of cancers such as proliferation, migration and response to hormones, suggesting that splicing-targeting therapy can be promising as a new strategy for cancer treatment. In this review, we focus on the splicing regulation by RNA-binding proteins including Drosophila behavior/human splicing (DBHS) family proteins, serine/arginine-rich (SR) proteins and heterogeneous nuclear ribonucleoproteins (hnRNPs) in hormone-related cancers, such as breast and prostate cancers. Keywords: DBHS family proteins; SR proteins; hnRNPs; breast cancer; prostate cancer 1. Introduction Splicing of mRNA precursors (pre-mRNAs) is an essential mechanism in the posttranscriptional regulation of gene expression. -

The Biological Function and Clinical Significance of SF3B1 Mutations In
Zhou et al. Biomarker Research (2020) 8:38 https://doi.org/10.1186/s40364-020-00220-5 REVIEW Open Access The biological function and clinical significance of SF3B1 mutations in cancer Zhixia Zhou1* , Qi Gong2, Yin Wang1, Mengkun Li1, Lu Wang1, Hongfei Ding1 and Peifeng Li1* Abstract Spliceosome mutations have become the most interesting mutations detected in human cancer in recent years. The spliceosome, a large, dynamic multimegadalton small nuclear ribonucleoprotein composed of small nuclear RNAs associated with proteins, is responsible for removing introns from precursor mRNA (premRNA) and generating mature, spliced mRNAs. SF3B1 is the largest subunit of the spliceosome factor 3b (SF3B) complex, which is a core component of spliceosomes. Recurrent somatic mutations in SF3B1 have been detected in human cancers, including hematological malignancies and solid tumors, and indicated to be related to patient prognosis. This review summarizes the research progress of SF3B1 mutations in cancer, including SF3B1 mutations in the HEAT domain, the multiple roles and aberrant splicing events of SF3B1 mutations in the pathogenesis of tumors, and changes in mutated cancer cells regarding sensitivity to SF3B small-molecule inhibitors. In addition, the potential of SF3B1 or its mutations to serve as biomarkers or therapeutic targets in cancer is discussed. The accumulated knowledge about SF3B1 mutations in cancer provides critical insight into the integral role the SF3B1 protein plays in mRNA splicing and suggests new targets for anticancer therapy. Keywords: SF3B1, Mutation, Cancer, RNA splicing Background human diseases, including cancer [5, 6]. Indeed, genome- Of the 3.3 billion base pairs of haploid DNA in the human wide studies have revealed more than 15,000 tumor- genome, approximately 20,000 protein-coding genes have associated splice variants in a wide variety of cancers [7, 8]. -

Titration of SF3B1 Activity Reveals Distinct Effects on the Transcriptome and Cell Physiology
International Journal of Molecular Sciences Article Titration of SF3B1 Activity Reveals Distinct Effects on the Transcriptome and Cell Physiology Karen S. Kim Guisbert * , Isiah Mossiah and Eric Guisbert Department of Biomedical and Chemical Engineering and Sciences, Florida Institute of Technology, Melbourne, FL 32937, USA; imossiah2016@my.fit.edu (I.M.); eguisbert@fit.edu (E.G.) * Correspondence: kkimguisbert@fit.edu Received: 19 November 2020; Accepted: 14 December 2020; Published: 17 December 2020 Abstract: SF3B1 is a core component of the U2 spliceosome that is frequently mutated in cancer. We have previously shown that titrating the activity of SF3B1, using the inhibitor pladienolide B (PB), affects distinct steps of the heat shock response (HSR). Here, we identify other genes that are sensitive to different levels of SF3B1 (5 vs. 100 nM PB) using RNA sequencing. Significant changes to mRNA splicing were identified at both low PB and high PB concentrations. Changes in expression were also identified in the absence of alternative splicing, suggesting that SF3B1 influences other gene expression pathways. Surprisingly, gene expression changes identified in low PB are not predictive of changes in high PB. Specific pathways were identified with differential sensitivity to PB concentration, including nonsense-mediated decay and protein-folding homeostasis, both of which were validated using independent reporter constructs. Strikingly, cells exposed to low PB displayed enhanced protein-folding capacity relative to untreated cells. These data reveal that the transcriptome is exquisitely sensitive to SF3B1 and suggests that the activity of SF3B1 is finely regulated to coordinate mRNA splicing, gene expression and cellular physiology. Keywords: SF3B1; heat shock response; HSR; alternative splicing; pladienolide B; nonsense-mediated decay; NMD; early transcription termination; proteostasis 1. -

Clinical Characterization and Prognostic Impact of SF3B1 and TP53 Gene Mutations in Myelodysplastic Syndromes
Universitätsklinikum Ulm Zentrum für Innere Medizin Klinik für Innere Medizin III Hämatologie, Onkologie, Palliativmedizin, Rheumatologie und Infektionskrankheiten Ärztlicher Direktor: Prof. Dr. med. H. Döhner Clinical characterization and prognostic impact of SF3B1 and TP53 gene mutations in myelodysplastic syndromes Dissertation zur Erlangung des Doktorgrades der Medizin der Medizinischen Fakultät der Universität Ulm Vorgelegt von Till Wallrabenstein, geb. in Münster 2017 Amtierender Dekan: Prof. Dr. rer. nat. Thomas Wirth 1. Berichterstatter: Prof. Dr. med. Konstanze Döhner 2. Berichterstatter: Prof. Dr. med. Ramin Lotfi Tag der Promotion: 25. Oktober 2018 Table of contents Abbreviations ......................................................................................................... III 1 Introduction ...................................................................................................... 1 1.1 MDS .................................................................................................................................... 1 1.1.1 Pathogenesis .............................................................................................................. 1 1.1.2 Standards of diagnosis and classification .................................................................. 2 1.1.3 Prognostic scores ....................................................................................................... 6 1.1.4 Therapy options ......................................................................................................... -

Expression Levels of SF3B3 Correlate with Prognosis and This Is the Author's Manuscript of Article Published in Final Edited Form As
endocrine resistance in estrogen receptor-positive breast cancer. Modern Pathology, 28(5), 677–685. http://doi.org/10.1038/modpathol.2014.146 Gökmen-Polar, Y., Neelamraju, Goswami, C. P., Gu, X., Nallamothu, G., Janga, S. C., & Badve, (2015). Expression levels of SF3B3 correlate with prognosis and This is the author's manuscript of article published in final edited form as: 1 Expression levels of SF3B3 correlate with prognosis and endocrine resistance in estrogen 2 receptor positive breast cancer 3 4 Yesim Gökmen-Polar1*, Yaseswini Neelamraju3, Chirayu P. Goswami4, Xiaoping Gu1, 5 Gouthami Nallamothu1, Sarath Chandra Janga3,4,5, and Sunil Badve1,2,6* 6 Departments of 1Pathology and Laboratory Medicine, 2Medicine, Indiana University School of 7 Medicine, Indianapolis, IN, 3Department of Biohealth Informatics, School of Informatics and 8 Computing, IUPUI, Indianapolis, IN, 4Center for Computational Biology and Bioinformatics, 9 Indiana University School of Medicine; 5Department of Medical and Molecular Genetics, 10 Indiana University School of Medicine; 6Indiana University Melvin and Bren Simon Cancer 11 Center, Indianapolis, IN. 12 13 Grant Support 14 This work was supported by the Susan G Komen for the Cure® and 100 Voices of Hope to Sunil 15 Badve. 16 17 Disclosure of Potential Conflicts of Interest 18 The authors disclose no potential conflicts of interest. 19 20 *Address of corresponding: 21 Yesim Gökmen-Polar, PhD 22 Department of Pathology and Laboratory Medicine 23 Indiana University School of Medicine 24 635 Barnhill Dr., MS 0038