The Biological Function and Clinical Significance of SF3B1 Mutations In
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Mutated SF3B1 Is Associated with Transcript Isoform Changes of The
bioRxiv preprint doi: https://doi.org/10.1101/000992; this version posted July 13, 2014. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Reyes et al. RESEARCH Mutated SF3B1 is associated with transcript isoform changes of the genes UQCC and RPL31 both in CLLs and uveal melanomas Alejandro Reyes1, Carolin Blume2, Vincent Pelechano1, Petra Jakob1, Lars M Steinmetz1,3, Thorsten Zenz2,4 and Wolfgang Huber1* *Correspondence: [email protected] 1European Molecular Biology Abstract Laboratory, Genome Biology Unit, 69117, Heidelberg Germany Background: Genome sequencing studies of chronic lympoid leukemia (CLL) Full list of author information is have provided a comprehensive overview of recurrent somatic mutations in coding available at the end of the article genes. One of the most intriguing discoveries has been the prevalence of mutations in the HEAT-repeat domain of the splicing factor SF3B1. A frequently observed variant is predicted to cause the substitution of a lysine with a glutamic acid at position 700 of the protein (K700E). However, the molecular consequences of the mutations are largely unknown. Results: To start exploring this question, we sequenced the transcriptomes of six samples: four samples of CLL tumour cells, of which two contained the K700E mutation in SF3B1, and CD19 positive cells from two healthy donors. We identified 41 genes that showed differential usage of exons statistically associated with the mutated status of SF3B1 (false discovery rate of 10%). -

SF3B3) and Sin3a Associated Protein 130 (SAP130
cells Communication Ambiguity about Splicing Factor 3b Subunit 3 (SF3B3) and Sin3A Associated Protein 130 (SAP130) Paula I. Metselaar 1,* , Celine Hos 1, Olaf Welting 1, Jos A. Bosch 2,3, Aletta D. Kraneveld 4 , Wouter J. de Jonge 1 and Anje A. Te Velde 1 1 Tytgat Institute for Liver and Intestinal Research, AGEM, Amsterdam UMC, University of Amsterdam, 1105BK Amsterdam, The Netherlands; [email protected] (C.H.); [email protected] (O.W.); [email protected] (W.J.d.J.); [email protected] (A.A.T.V.) 2 Department of Psychology, University of Amsterdam, 1018WS Amsterdam, The Netherlands; [email protected] 3 Department of Medical Psychology, Amsterdam UMC, University of Amsterdam, 1001NK Amsterdam, The Netherlands 4 Division of Pharmacology, Utrecht Institute for Pharmaceutical Sciences, Faculty of Science, Utrecht University, 3584CG Utrecht, The Netherlands; [email protected] * Correspondence: [email protected] Abstract: In 2020, three articles were published on a protein that can activate the immune system by binding to macrophage-inducible C-type lectin receptor (Mincle). In the articles, the protein was referred to as ‘SAP130, a subunit of the histone deacetylase complex.’ However, the Mincle ligand the authors aimed to investigate is splicing factor 3b subunit 3 (SF3B3). This splicing factor is unrelated to SAP130 (Sin3A associated protein 130, a subunit of the histone deacetylase-dependent Sin3A corepressor complex). The conclusions in the three articles were formulated for SF3B3, Citation: Metselaar, P.I.; Hos, C.; while the researchers used qPCR primers and antibodies against SAP130. -

Anti-Phospho-SF3B1 (Sap155) (Ser129) Pab
PD043 Page 1 For Research Use Only. Not for use in diagnostic procedures. Anti-Phospho-SF3B1 (Sap155) (Ser129) pAb CODE No. PD043 CLONALITY Polyclonal ISOTYPE Rabbit Ig, affinity purified QUANTITY 100 L SOURCE Purified IgG from rabbit serum IMMUNOGEN KLH conjugated synthetic peptide, RTMII(pS)PERL (corresponding to amino acid residues 124-133 of human SF3B1) FORMURATION PBS containing 50% Glycerol (pH 7.2). No preservative is contained. STORAGE This antibody solution is stable for one year from the date of purchase when stored at -20°C. APPLICATION-CONFIRMED Western blotting 1:1000 for chemiluminescence detection system SPECIES CROSS REACTIVITY on WB Species Human Mouse Rat Hamster Cell HeLa, Raji NIH/3T3, WR19L Not tested Not tested Reactivity Entrez Gene ID 23451 (Human), 81898 (Mouse) REFERENCES 1) Wang, C., et al., Genes Dev. 12, 1409-1414 (1998) 2) Shi, Y., et al., Mol Cell 23, 819-829 (2006) 3) Tanuma, N., et al., J. Biol. Chem., 283, 35805-35814 (2008) 4) Yoshida, K., et al., Nature 478, 64-69 (2011) 5) Rossi, D., et al., Blood 118, 6904-6908 (2011) 6) Quesada, V., et al., Nat Genet 44, 47-52 (2011) 7) Wang, L., et al., N Engl J Med. 365, 2497-2506 (2011) 8) Biankin, A. V., et al., Nature 491, 399-405 (2012) For more information, please visit our web site http://ruo.mbl.co.jp/ MEDICAL & BIOLOGICAL LABORATORIES CO., LTD. URL http://ruo.mbl.co.jp/ e-mail [email protected], TEL 052-238-1904 PD043 Page 2 P a g RELATED PRODUCTS e PD043 Anti-Phospho-SF3B1 (Sap155) (Ser129) pAb D138-3 Anti-Sap155 (SF3B12) mAb D221-3 Anti-Sap155 (SF3B1) -

Monoclonal B-Cell Lymphocytosis Is Characterized by Mutations in CLL Putative Driver Genes and Clonal Heterogeneity Many Years Before Disease Progression
Leukemia (2014) 28, 2395–2424 © 2014 Macmillan Publishers Limited All rights reserved 0887-6924/14 www.nature.com/leu LETTERS TO THE EDITOR Monoclonal B-cell lymphocytosis is characterized by mutations in CLL putative driver genes and clonal heterogeneity many years before disease progression Leukemia (2014) 28, 2395–2398; doi:10.1038/leu.2014.226 (Beckton Dickinson) and data analyzed using Cell Quest software. On the basis of FACS (fluorescence-activated cell sorting) analysis, we observed after enrichment an average of 91% of CD19+ cells Monoclonal B-cell lymphocytosis (MBL) is defined as an asympto- (range 76–99%) and 91% of the CD19+ fraction were CD19+/CD5+ matic expansion of clonal B cells with less than 5 × 109/L cells in the cells (range 66–99%). We used the values of the CD19+/CD5+ peripheral blood and without other manifestations of chronic fraction to calculate the leukemic B-cell fraction and reduce any lymphocytic leukemia (CLL; for example, lymphadenopathy, cyto- significant contamination of non-clonal B cells in each biopsy. DNA penias, constitutional symptoms).1 Approximately 1% of the MBL was extracted from the clonal B cells and non-clonal (that is, T cells) cohort develops CLL per year. Evidence suggests that nearly all CLL cells using the Gentra Puregene Cell Kit (Qiagen, Hilden, Germany). 2 fi fi cases are preceded by an MBL state. Our understanding of the Extracted DNAs were ngerprinted to con rm the relationship genetic basis, clonal architecture and evolution in CLL pathogenesis between samples of the same MBL individual and to rule out sample has undergone significant improvements in the last few years.3–8 In cross-contamination between individuals. -

SF3B2-Mediated RNA Splicing Drives Human Prostate Cancer Progression
Published OnlineFirst August 20, 2019; DOI: 10.1158/0008-5472.CAN-18-3965 Cancer Molecular Cell Biology Research SF3B2-Mediated RNA Splicing Drives Human Prostate Cancer Progression Norihiko Kawamura1,2, Keisuke Nimura1, Kotaro Saga1, Airi Ishibashi1, Koji Kitamura1,3, Hiromichi Nagano1, Yusuke Yoshikawa4, Kyoso Ishida1,5, Norio Nonomura2, Mitsuhiro Arisawa4, Jun Luo6, and Yasufumi Kaneda1 Abstract Androgen receptor splice variant-7 (AR-V7) is a General RNA splicing SF3B2 complex-mediated alternative RNA splicing constitutively active AR variant implicated in U2 castration-resistant prostate cancers. Here, we show U2 snRNA that the RNA splicing factor SF3B2, identified by 3’ 3’ in silico and CRISPR/Cas9 analyses, is a critical 5’ 3’ splice site 5’ SF3B7 AR-V7 5’ A U2AF2 AGA Exon ? determinant of expression and is correlated SF3B6(p14) SF3B4 SF3B1 SF3B4 SF3B1 with aggressive cancer phenotypes. Transcriptome SF3B5 SF3B2 SF3B3 SF3B2 SF3B3 and PAR-CLIP analyses revealed that SF3B2 con- SF3A3 SF3B2 complex SF3A3 SF3A1 SF3A1 SF3b complex trols the splicing of target genes, including AR, to AR pre-mRNA drive aggressive phenotypes. SF3B2-mediated CE3 aggressive phenotypes in vivo were reversed by AR-V7 mRNA AR mRNA AR-V7 knockout. Pladienolide B, an inhibitor of CE3 a splicing modulator of the SF3b complex, sup- Drive malignancy pressed the growth of tumors addicted to high While the SF3b complex is critical for general RNA splicing, SF3B2 promotes inclusion of the target exon through recognizing a specific RNA motif. SF3B2 expression. These findings support the idea © 2019 American Association for Cancer Research that alteration of the splicing pattern by high SF3B2 expression is one mechanism underlying prostate cancer progression and therapeutic resistance. -

Flexible, Unbiased Analysis of Biological Characteristics Associated with Genomic Regions
bioRxiv preprint doi: https://doi.org/10.1101/279612; this version posted March 22, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. BioFeatureFinder: Flexible, unbiased analysis of biological characteristics associated with genomic regions Felipe E. Ciamponi 1,2,3; Michael T. Lovci 2; Pedro R. S. Cruz 1,2; Katlin B. Massirer *,1,2 1. Structural Genomics Consortium - SGC, University of Campinas, SP, Brazil. 2. Center for Molecular Biology and Genetic Engineering - CBMEG, University of Campinas, Campinas, SP, Brazil. 3. Graduate program in Genetics and Molecular Biology, PGGBM, University of Campinas, Campinas, SP, Brazil. *Corresponding author: [email protected] Mailing address: Center for Molecular Biology and Genetic Engineering - CBMEG, University of Campinas, Campinas, SP, Brazil. Av Candido Rondo, 400 Cidade Universitária CEP 13083-875, Campinas, SP Phone: 55-19-98121-937 bioRxiv preprint doi: https://doi.org/10.1101/279612; this version posted March 22, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Abstract BioFeatureFinder is a novel algorithm which allows analyses of many biological genomic landmarks (including alternatively spliced exons, DNA/RNA- binding protein binding sites, and gene/transcript functional elements, nucleotide content, conservation, k-mers, secondary structure) to identify distinguishing features. -

Full-Length Transcript Characterization of SF3B1 Mutation in Chronic Lymphocytic Leukemia Reveals Downregulation of Retained Introns
ARTICLE https://doi.org/10.1038/s41467-020-15171-6 OPEN Full-length transcript characterization of SF3B1 mutation in chronic lymphocytic leukemia reveals downregulation of retained introns Alison D. Tang 1, Cameron M. Soulette 2, Marijke J. van Baren1, Kevyn Hart1, Eva Hrabeta-Robinson1, ✉ Catherine J. Wu 3,4,5 & Angela N. Brooks 1 SF3B1 1234567890():,; While splicing changes caused by somatic mutations in are known, identifying full- length isoform changes may better elucidate the functional consequences of these mutations. We report nanopore sequencing of full-length cDNA from CLL samples with and without SF3B1 mutation, as well as normal B cell samples, giving a total of 149 million pass reads. We present FLAIR (Full-Length Alternative Isoform analysis of RNA), a computational workflow to identify high-confidence transcripts, perform differential splicing event analysis, and dif- ferential isoform analysis. Using nanopore reads, we demonstrate differential 3’ splice site changes associated with SF3B1 mutation, agreeing with previous studies. We also observe a strong downregulation of intron retention events associated with SF3B1 mutation. Full-length transcript analysis links multiple alternative splicing events together and allows for better estimates of the abundance of productive versus unproductive isoforms. Our work demon- strates the potential utility of nanopore sequencing for cancer and splicing research. 1 Department of Biomolecular Engineering, University of California, Santa Cruz, CA 95062, USA. 2 Department of Molecular Cell & Developmental Biology, University of California, Santa Cruz, CA 95062, USA. 3 Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, MA 02215, USA. 4 Broad Institiute of Harvard and MIT, Cambridge, MA, USA. -

Open Data for Differential Network Analysis in Glioma
International Journal of Molecular Sciences Article Open Data for Differential Network Analysis in Glioma , Claire Jean-Quartier * y , Fleur Jeanquartier y and Andreas Holzinger Holzinger Group HCI-KDD, Institute for Medical Informatics, Statistics and Documentation, Medical University Graz, Auenbruggerplatz 2/V, 8036 Graz, Austria; [email protected] (F.J.); [email protected] (A.H.) * Correspondence: [email protected] These authors contributed equally to this work. y Received: 27 October 2019; Accepted: 3 January 2020; Published: 15 January 2020 Abstract: The complexity of cancer diseases demands bioinformatic techniques and translational research based on big data and personalized medicine. Open data enables researchers to accelerate cancer studies, save resources and foster collaboration. Several tools and programming approaches are available for analyzing data, including annotation, clustering, comparison and extrapolation, merging, enrichment, functional association and statistics. We exploit openly available data via cancer gene expression analysis, we apply refinement as well as enrichment analysis via gene ontology and conclude with graph-based visualization of involved protein interaction networks as a basis for signaling. The different databases allowed for the construction of huge networks or specified ones consisting of high-confidence interactions only. Several genes associated to glioma were isolated via a network analysis from top hub nodes as well as from an outlier analysis. The latter approach highlights a mitogen-activated protein kinase next to a member of histondeacetylases and a protein phosphatase as genes uncommonly associated with glioma. Cluster analysis from top hub nodes lists several identified glioma-associated gene products to function within protein complexes, including epidermal growth factors as well as cell cycle proteins or RAS proto-oncogenes. -

Mutations in Splicing Factor Genes in Myeloid Malignancies: Significance and Impact on Clinical Features
cancers Review Mutations in Splicing Factor Genes in Myeloid Malignancies: Significance and Impact on Clinical Features Valeria Visconte 1, Megan O. Nakashima 2 and Heesun J. Rogers 2,* 1 Department of Translational Hematology and Oncology Research, Taussig Cancer Institute, Cleveland Clinic, Cleveland, OH 44195, USA; [email protected] 2 Department of Laboratory Medicine, Cleveland Clinic, Cleveland, OH 44195, USA; [email protected] * Correspondence: [email protected]; Tel.: +1-216-445-2719 Received: 15 October 2019; Accepted: 19 November 2019; Published: 22 November 2019 Abstract: Components of the pre-messenger RNA splicing machinery are frequently mutated in myeloid malignancies. Mutations in LUC7L2, PRPF8, SF3B1, SRSF2, U2AF1, and ZRSR2 genes occur at various frequencies ranging between 40% and 85% in different subtypes of myelodysplastic syndrome (MDS) and 5% and 10% of acute myeloid leukemia (AML) and myeloproliferative neoplasms (MPNs). In some instances, splicing factor (SF) mutations have provided diagnostic utility and information on clinical outcomes as exemplified by SF3B1 mutations associated with increased ring sideroblasts (RS) in MDS-RS or MDS/MPN-RS with thrombocytosis. SF3B1 mutations are associated with better survival outcomes, while SRSF2 mutations are associated with a shorter survival time and increased AML progression, and U2AF1 mutations with a lower remission rate and shorter survival time. Beside the presence of mutations, transcriptomics technologies have shown that one third of genes in AML patients are differentially expressed, leading to altered transcript stability, interruption of protein function, and improper translation compared to those of healthy individuals. The detection of SF mutations demonstrates the importance of splicing abnormalities in the hematopoiesis of MDS and AML patients given the fact that abnormal splicing regulates the function of several transcriptional factors (PU.1, RUNX1, etc.) crucial in hematopoietic function. -
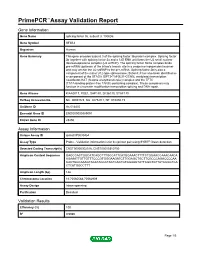
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name splicing factor 3b, subunit 3, 130kDa Gene Symbol SF3B3 Organism Human Gene Summary This gene encodes subunit 3 of the splicing factor 3b protein complex. Splicing factor 3b together with splicing factor 3a and a 12S RNA unit forms the U2 small nuclear ribonucleoproteins complex (U2 snRNP). The splicing factor 3b/3a complex binds pre-mRNA upstream of the intron's branch site in a sequence independent manner and may anchor the U2 snRNP to the pre-mRNA. Splicing factor 3b is also a component of the minor U12-type spliceosome. Subunit 3 has also been identified as a component of the STAGA (SPT3-TAF(II)31-GCN5L acetylase) transcription coactivator-HAT (histone acetyltransferase) complex and the TFTC (TATA-binding-protein-free TAF(II)-containing complex). These complexes may function in chromatin modification transcription splicing and DNA repair. Gene Aliases KIAA0017, RSE1, SAP130, SF3b130, STAF130 RefSeq Accession No. NC_000016.9, NG_027529.1, NT_010498.15 UniGene ID Hs.514435 Ensembl Gene ID ENSG00000189091 Entrez Gene ID 23450 Assay Information Unique Assay ID qHsaCIP0030454 Assay Type Probe - Validation information is for the primer pair using SYBR® Green detection Detected Coding Transcript(s) ENST00000302516, ENST00000310750 Amplicon Context Sequence GAGCCACTGGCATCAGCTTTGCCATTCATGGAAACTTTTCTGGAACCAAACAACA AGAAATTGTTGTTTCCCGTGGGAAGATCTTGGAGCTGCTTCGCCCAGACCCCAA CACTGGCAAAGTACATACCCTACTCACTGTGGAAGTATTCGGTGTTATCCGGTCA CTCATGGCCTTT Amplicon Length (bp) 146 Chromosome Location 16:70560588-70562909 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 100 R2 0.9986 Page 1/5 PrimePCR™Assay Validation Report cDNA Cq 18.72 cDNA Tm (Celsius) 83 gDNA Cq Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. -
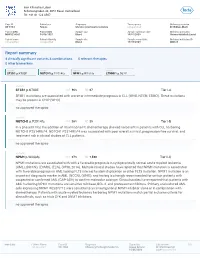
Report Summary 4 Clinically Significant Variants & Combinations 0 Relevant Therapies 0 Other Biomarkers
Inov Klinisches Labor Schützengraben 42, 4051 Basel, Switzerland Tel: +41 61 123 4567 Case ID Patient sex Diagnosis Tumor purity Ordering physician XB-1064 Female Chronic lymphocytic leukemia not specified Dr. Niklaus Marti Patient MRN Patient DOB Sample type Sample collection date Ordering institution MRN1234567 04/08/1957 Blood 10/01/2020 Universitätsklinik Liestal Patient name Patient ethnicity Sample site Sample receipt date Ordering institution ID not specified not specified Blood 10/03/2020 OID123 Report summary 4 clinically significant variants & combinations 0 relevant therapies 0 other biomarkers variant variant variant variant SF3B1 p.K700E NOTCH1 p.P2514fs NPM1 p.W288fs CTNNB1 p.D32Y variant SF3B1 p.K700E VAF 36% RD 67 Tier I-A SF3B1 mutations are associated with worse or intermediate prognosis in CLL (WHO, NCCN, ESMO). These mutations may be present in CHIP (WHO). no approved therapies variant NOTCH1 p.P2514fs VAF 28% RD 25 Tier I-B In a phase III trial, the addition of rituximab to FC chemotherapy showed no benefit in patients with CLL harboring NOTCH1 P2514Rfs*4. NOTCH1 P2514Rfs*4 was associated with poor overall survival, progression-free survival, and treatment risk in clinical studies of CLL patients. no approved therapies variant NPM1 p.W288fs VAF 27% RD 1,590 Tier II-C NPM1 mutations are associated with with a favorable prognosis in cytogenetically normal acute myeloid leukemia (AML), (NCCN), (ESMO), (ELN), (WHO, 2016). Multiple clinical studies have reported that NPM1 mutation is associated with favorable prognosis in AML lacking FLT3 internal tandem duplication or other FLT3 mutation. NPM1 mutation is an important diagnostic marker in AML (NCCN), (WHO), and testing is strongly recommended for certain patients with suspected or confirmed AML (CAP-ASH) to confirm molecular subtype. -

A High-Throughput Approach to Uncover Novel Roles of APOBEC2, a Functional Orphan of the AID/APOBEC Family
Rockefeller University Digital Commons @ RU Student Theses and Dissertations 2018 A High-Throughput Approach to Uncover Novel Roles of APOBEC2, a Functional Orphan of the AID/APOBEC Family Linda Molla Follow this and additional works at: https://digitalcommons.rockefeller.edu/ student_theses_and_dissertations Part of the Life Sciences Commons A HIGH-THROUGHPUT APPROACH TO UNCOVER NOVEL ROLES OF APOBEC2, A FUNCTIONAL ORPHAN OF THE AID/APOBEC FAMILY A Thesis Presented to the Faculty of The Rockefeller University in Partial Fulfillment of the Requirements for the degree of Doctor of Philosophy by Linda Molla June 2018 © Copyright by Linda Molla 2018 A HIGH-THROUGHPUT APPROACH TO UNCOVER NOVEL ROLES OF APOBEC2, A FUNCTIONAL ORPHAN OF THE AID/APOBEC FAMILY Linda Molla, Ph.D. The Rockefeller University 2018 APOBEC2 is a member of the AID/APOBEC cytidine deaminase family of proteins. Unlike most of AID/APOBEC, however, APOBEC2’s function remains elusive. Previous research has implicated APOBEC2 in diverse organisms and cellular processes such as muscle biology (in Mus musculus), regeneration (in Danio rerio), and development (in Xenopus laevis). APOBEC2 has also been implicated in cancer. However the enzymatic activity, substrate or physiological target(s) of APOBEC2 are unknown. For this thesis, I have combined Next Generation Sequencing (NGS) techniques with state-of-the-art molecular biology to determine the physiological targets of APOBEC2. Using a cell culture muscle differentiation system, and RNA sequencing (RNA-Seq) by polyA capture, I demonstrated that unlike the AID/APOBEC family member APOBEC1, APOBEC2 is not an RNA editor. Using the same system combined with enhanced Reduced Representation Bisulfite Sequencing (eRRBS) analyses I showed that, unlike the AID/APOBEC family member AID, APOBEC2 does not act as a 5-methyl-C deaminase.