Genome-Wide Transcriptome Profiling of Ex-Vivo Precision-Cut Slices From
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-
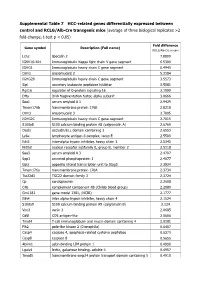
Supplemental Table 7 HCC-Related Genes Differentially Expressed Between Control and RCLG/Alb-Cre Transgenic Mice
Supplemental Table 7 HCC-related genes differentially expressed between control and RCLG/Alb-Cre transgenic mice (average of three biological replicates >2 fold-change, t-test p < 0.05) Fold difference Gene symbol Description (Full name) (RCLG/Alb-Cre vs con) Lcn2 lipocalin 2 7.8899 IGKV16-104 Immunoglobulin Kappa light chain V gene segment 6.5300 IGHG1 Immunoglobulin heavy chain C gene segment 6.4945 Orm2 orosomucoid 2 5.3184 IGHG2B Immunoglobulin heavy chain C gene segment 3.5573 Slpi secretory leukocyte peptidase inhibitor 3.5081 Rgs16 regulator of G-protein signaling 16 3.1999 Dffa DNA fragmentation factor, alpha subunit 3.0666 Saa1 serum amyloid A 1 2.9429 Tmem176b transmembrane protein 176B 2.8218 Orm3 orosomucoid 3 2.7805 IGHG2C Immunoglobulin heavy chain C gene segment 2.7015 S100a8 S100 calcium binding protein A8 (calgranulin A) 2.6769 Ocel1 occludin/ELL domain containing 1 2.6553 Ly6e lymphocyte antigen 6 complex, locus E 2.5509 Itih3 inter-alpha trypsin inhibitor, heavy chain 3 2.5345 Nr0b2 nuclear receptor subfamily 0, group B, member 2 2.5118 Saa3 serum amyloid A 3 2.4707 Spp1 secreted phosphoprotein 1 2.4077 Gats opposite strand transcription unit to Stag3 2.3934 Tmem176a transmembrane protein 176A 2.3734 Tsc22d3 TSC22 domain family 3 2.3724 Cp ceruloplasmin 2.2608 C4b complement component 4B (Childo blood group) 2.2089 Gm1381 gene model 1381, (NCBI) 2.1777 Itih4 inter alpha-trypsin inhibitor, heavy chain 4 2.1524 S100a9 S100 calcium binding protein A9 (calgranulin B) 2.124 Vnn3 vanin 3 2.0685 Cd5l CD5 antigen-like 2.0606 -

4-6 Weeks Old Female C57BL/6 Mice Obtained from Jackson Labs Were Used for Cell Isolation
Methods Mice: 4-6 weeks old female C57BL/6 mice obtained from Jackson labs were used for cell isolation. Female Foxp3-IRES-GFP reporter mice (1), backcrossed to B6/C57 background for 10 generations, were used for the isolation of naïve CD4 and naïve CD8 cells for the RNAseq experiments. The mice were housed in pathogen-free animal facility in the La Jolla Institute for Allergy and Immunology and were used according to protocols approved by the Institutional Animal Care and use Committee. Preparation of cells: Subsets of thymocytes were isolated by cell sorting as previously described (2), after cell surface staining using CD4 (GK1.5), CD8 (53-6.7), CD3ε (145- 2C11), CD24 (M1/69) (all from Biolegend). DP cells: CD4+CD8 int/hi; CD4 SP cells: CD4CD3 hi, CD24 int/lo; CD8 SP cells: CD8 int/hi CD4 CD3 hi, CD24 int/lo (Fig S2). Peripheral subsets were isolated after pooling spleen and lymph nodes. T cells were enriched by negative isolation using Dynabeads (Dynabeads untouched mouse T cells, 11413D, Invitrogen). After surface staining for CD4 (GK1.5), CD8 (53-6.7), CD62L (MEL-14), CD25 (PC61) and CD44 (IM7), naïve CD4+CD62L hiCD25-CD44lo and naïve CD8+CD62L hiCD25-CD44lo were obtained by sorting (BD FACS Aria). Additionally, for the RNAseq experiments, CD4 and CD8 naïve cells were isolated by sorting T cells from the Foxp3- IRES-GFP mice: CD4+CD62LhiCD25–CD44lo GFP(FOXP3)– and CD8+CD62LhiCD25– CD44lo GFP(FOXP3)– (antibodies were from Biolegend). In some cases, naïve CD4 cells were cultured in vitro under Th1 or Th2 polarizing conditions (3, 4). -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

The Roles of RNA Polymerase I and III Subunits Polr1a, Polr1c, and Polr1d in Craniofacial Development BY
The roles of RNA Polymerase I and III subunits Polr1a, Polr1c, and Polr1d in craniofacial development BY © 2016 Kristin Emily Noack Watt Submitted to the graduate degree program in Anatomy and Cell Biology and to the Graduate Faculty of The University of Kansas Medical Center in partial fulfillment of the requirements for the degree of Doctor of Philosophy. Paul Trainor, Co-Chairperson Brenda Rongish, Co-Chairperson Brian Andrews Jennifer Gerton Tatjana Piotrowski Russell Swerdlow Date Defended: January 26, 2016 The Dissertation Committee for Kristin Watt certifies that this is the approved version of the following dissertation: The roles of RNA Polymerase I and III subunits Polr1a, Polr1c, and Polr1d in craniofacial development Paul Trainor, Co-Chairperson Brenda Rongish, Co-Chairperson Date approved: February 2, 2016 ii Abstract Craniofacial anomalies account for approximately one-third of all birth defects. Two examples of syndromes associated with craniofacial malformations are Treacher Collins syndrome and Acrofacial Dysostosis, Cincinnati type which have phenotypic overlap including deformities of the eyes, ears, and facial bones. Mutations in TCOF1, POLR1C or POLR1D may cause Treacher Collins syndrome while mutations in POLR1A may cause Acrofacial Dysostosis, Cincinnati type. TCOF1 encodes the nucleolar phosphoprotein Treacle, which functions in rRNA transcription and modification. Previous studies demonstrated that Tcof1 mutations in mice result in reduced ribosome biogenesis and increased neuroepithelial apoptosis. This diminishes the neural crest cell (NCC) progenitor population which contribute to the development of the cranial skeleton. In contrast, apart from being subunits of RNA Polymerases (RNAP) I and/or III, nothing is known about the function of POLR1A, POLR1C, and POLR1D during embryonic and craniofacial development. -

MOCHI Enables Discovery of Heterogeneous Interactome Modules in 3D Nucleome
Downloaded from genome.cshlp.org on October 4, 2021 - Published by Cold Spring Harbor Laboratory Press MOCHI enables discovery of heterogeneous interactome modules in 3D nucleome Dechao Tian1,# , Ruochi Zhang1,# , Yang Zhang1, Xiaopeng Zhu1, and Jian Ma1,* 1Computational Biology Department, School of Computer Science, Carnegie Mellon University, Pittsburgh, PA 15213, USA #These two authors contributed equally *Correspondence: [email protected] Contact To whom correspondence should be addressed: Jian Ma School of Computer Science Carnegie Mellon University 7705 Gates-Hillman Complex 5000 Forbes Avenue Pittsburgh, PA 15213 Phone: +1 (412) 268-2776 Email: [email protected] 1 Downloaded from genome.cshlp.org on October 4, 2021 - Published by Cold Spring Harbor Laboratory Press Abstract The composition of the cell nucleus is highly heterogeneous, with different constituents forming complex interactomes. However, the global patterns of these interwoven heterogeneous interactomes remain poorly understood. Here we focus on two different interactomes, chromatin interaction network and gene regulatory network, as a proof-of-principle, to identify heterogeneous interactome modules (HIMs), each of which represents a cluster of gene loci that are in spatial contact more frequently than expected and that are regulated by the same group of transcription factors. HIM integrates transcription factor binding and 3D genome structure to reflect “transcriptional niche” in the nucleus. We develop a new algorithm MOCHI to facilitate the discovery of HIMs based on network motif clustering in heterogeneous interactomes. By applying MOCHI to five different cell types, we found that HIMs have strong spatial preference within the nucleus and exhibit distinct functional properties. Through integrative analysis, this work demonstrates the utility of MOCHI to identify HIMs, which may provide new perspectives on the interplay between transcriptional regulation and 3D genome organization. -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in -

The Genetic Basis of Malformation of Cortical Development Syndromes: Primary Focus on Aicardi Syndrome
The Genetic Basis of Malformation of Cortical Development Syndromes: Primary Focus on Aicardi Syndrome Thuong Thi Ha B. Sc, M. Bio Neurogenetics Research Group The University of Adelaide Thesis submitted for the degree of Doctor of Philosophy In Discipline of Genetics and Evolution School of Biological Sciences Faculty of Science The University of Adelaide June 2018 Table of Contents Abstract 6 Thesis Declaration 8 Acknowledgements 9 Publications 11 Abbreviations 12 CHAPTER I: Introduction 15 1.1 Overview of Malformations of Cortical Development (MCD) 15 1.2 Introduction into Aicardi Syndrome 16 1.3 Clinical Features of Aicardi Syndrome 20 1.3.1 Epidemiology 20 1.3.2 Clinical Diagnosis 21 1.3.3 Differential Diagnosis 23 1.3.4 Development & Prognosis 24 1.4 Treatment 28 1.5 Pathogenesis of Aicardi Syndrome 29 1.5.1 Prenatal or Intrauterine Disturbances 29 1.5.2 Genetic Predisposition 30 1.6 Hypothesis & Aims 41 1.6.1 Hypothesis 41 1.6.2 Research Aims 41 1.7 Expected Outcomes 42 CHAPTER II: Materials & Methods 43 2.1 Study Design 43 2.1.1 Cohort of Study 44 2.1.2 Inheritance-based Strategy 45 1 2.1.3 Ethics for human and animals 46 2.2 Computational Methods 46 2.2.1 Pre-Processing Raw Reads 46 2.2.2 Sequencing Coverage 47 2.2.3 Variant Discovery 49 2.2.4 Annotating Variants 53 2.2.5 Evaluating Variants 55 2.3 Biological Methods 58 2.3.1 Cell Culture 58 2.3.2 Genomic DNA Sequencing 61 2.3.3 Plasmid cloning 69 2.3.4 RNA, whole exome & Whole Genome Sequencing 74 2.3.5 TOPFlash Assay 75 2.3.6 Western Blot 76 2.3.7 Morpholino Knockdown in Zebrafish 79 CHAPTER III: A mutation in COL4A2 causes autosomal dominant porencephaly with cataracts. -

Mutant P53 Uses P63 As a Molecular Chaperone to Alter Gene Expression and Induce a Pro-Invasive Secretome
www.impactjournals.com/oncotarget/ Oncotarget, December, Vol.2, No 12 Mutant p53 uses p63 as a molecular chaperone to alter gene expression and induce a pro-invasive secretome Paul M. Neilsen1,*, Jacqueline E. Noll1,*, Rachel J. Suetani1, Renee B. Schulz1, Fares Al-Ejeh2, Andreas Evdokiou3, David P. Lane4 and David F. Callen1 1 Cancer Therapeutics Laboratory, Discipline of Medicine, University of Adelaide, Australia 2 Signal Transduction Laboratory, Queensland Institute for Medical Research, Australia 3 Discipline of Surgery, Basil Hetzel Institute, University of Adelaide, Australia 4 p53Lab, Immunos, Agency for Science, Technology and Research, Singapore * Denotes Equal Contribution Correspondence to: Paul Neilsen, email: [email protected] Keywords: mutant p53, p63, secretome, invasion Received: December 9, 2011, Accepted: December 23, 2011, Published: December 25, 2011 Copyright: © Neilsen et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT: Mutations in the TP53 gene commonly result in the expression of a full-length protein that drives cancer cell invasion and metastasis. Herein, we have deciphered the global landscape of transcriptional regulation by mutant p53 through the application of a panel of isogenic H1299 derivatives with inducible expression of several common cancer-associated p53 mutants. We found that the ability of mutant p53 to alter the transcriptional profile of cancer cells is remarkably conserved across different p53 mutants. The mutant p53 transcriptional landscape was nested within a small subset of wild-type p53 responsive genes, suggesting that the oncogenic properties of mutant p53 are conferred by retaining its ability to regulate a defined set of p53 target genes. -

View Association Domain Familymember9(RASSF9);(6) • No Space Constraints Or Color figure Charges Occludin/ELL Domain Containing 1 (OCEL1)
Xing et al. BMC Proceedings 2011, 5(Suppl 1):P104 http://www.biomedcentral.com/1753-6561/5/S1/P104 POSTER PRESENTATION Open Access Screening and identification of host cellular factors interaction with immediate-early protein ICP22 of herpes simplex virus type 1 Junji Xing*, Fusen Lin, Meili Li, Shuai Wang, Hong Guo, Chunfu Zheng From Institut Pasteur International Network Annual Scientific Meeting Hong Kong. 22-23 November 2010 Herpes simplex virus type 1 (HSV-1) is a common and enhanced cyan fluorescent protein (ECFP), enhanced widely studied human pathogen that can replicate in yellow fluorescent protein (EYFP), respectively. Upon epithelial cells and other cells of the host or alternatively cotransfection of COS-7 cells, RASSF9-EYFP and can remain latent in peripheral neurons. ICP22 consists OCEL1-EYFP both co-localized with ICP22-ECFP in dis- of 420 residues and is encoded by a spliced mRNA tran- tinct nuclear domains, indicating they are host cellular scribed from the US1 gene. It is necessary for efficient factors interaction with viral ICP22 under physiological HSV-1 growth in animal models of infection as well as conditions. for efficient in vitro growth in some, but not all, cul- tured cells. For example, ICP22 mutants grow well in Published: 10 January 2011 African green monkey kidney (Vero) cells, but not in human embryonic lung (HEL) cells. ICP22 is extensively phosphorylated during infection, primarily by UL13 and doi:10.1186/1753-6561-5-S1-P104 another viral protein kinase, US3. In addition to indu- Cite this article as: Xing et al.: Screening and identification of host cing the modification of the host cell RNA Pol II, sev- cellular factors interaction with immediate-early protein ICP22 of herpes eral other functions have been attributed to ICP22; simplex virus type 1. -

Functional Effects of Variation in Transcription Factor Binding Highlight Long-Range Gene Regulation by Epromoters
bioRxiv preprint doi: https://doi.org/10.1101/620062; this version posted April 29, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Functional effects of variation in transcription factor binding highlight long-range gene regulation by epromoters Joanna Mitchelmore1, Nastasiya Grinberg2, Chris Wallace2,3 and Mikhail Spivakov1,4,5,* 1 Nuclear Dynamics Programme, Babraham Institute, Babraham Research Campus, Cambridge CB22 3AT, UK 2 Cambridge Institute of Therapeutic Immunology & Infectious Disease (CITIID), Cambridge Biomedical Campus, University of Cambridge, Cambridge, CB2 0AW, UK 3 MRC Biostatistics Unit, Cambridge Biomedical Campus, University of Cambridge, Cambridge CB2 0SR, UK 4 MRC London Institute of Medical Sciences, Du Cane Road, London W12 0NN, UK 5 Institute of Clinical Sciences, Faculty of Medicine, Imperial College, Du Cane Road, London W12 0NN, UK * Corresponding author. Email [email protected] Present address: Joanna Mitchelmore, Novartis Institutes of Biomedical Research, Fabrikstrasse 16, Novartis Campus Basel, 4056. Switzerland ABSTRACT Identifying DNA cis-regulatory modules (CRMs) that control the expression of specific genes is crucial for deciphering the logic of transcriptional control. Natural genetic variation can point to the possible gene regulatory function of specific sequences through their allelic associations with gene expression. However, comprehensive identification of causal regulatory sequences in brute- force association testing without incorporating prior knowledge is challenging due to limited statistical power and effects of linkage disequilibrium. Sequence variants affecting transcription factor (TF) binding at CRMs have a strong potential to influence gene regulatory function, which provides a motivation for prioritising such variants in association testing. -

(B6;129.Cg-Gt(ROSA)26Sor Tm20(CAG-Ctgf-GFP)Jsd) Were Crossed with Female Foxd1cre/+ Heterozygote Mice 1, and Experimental Mice Were Selected As Foxd1cre/+; Rs26cig/+
Supplemental Information SI Methods Animal studies Heterozygote mice (B6;129.Cg-Gt(ROSA)26Sor tm20(CAG-Ctgf-GFP)Jsd) were crossed with female Foxd1Cre/+ heterozygote mice 1, and experimental mice were selected as Foxd1Cre/+; Rs26CIG/+. In some studies Coll-GFPTg or TCF/Lef:H2B-GFPTg mice or Foxd1Cre/+; Rs26tdTomatoR/+ mice were used as described 2; 3. Left kidneys were subjected to ureteral obstruction using a posterior surgical approach as described 2. In some experiments recombinant mouse DKK1 (0.5mg/kg) or an equal volume of vehicle was administered by daily IP injection. In the in vivo ASO experiment, either specific Lrp6 (TACCTCAATGCGATTT) or scrambled negative control ASO (AACACGTCTATACGC) (30mg/kg) (Exiqon, LNA gapmers) was administered by IP injection on d-1, d1, d4, and d7. In other experiments anti-CTGF domain-IV antibodies (5mg/kg) or control IgG were administered d-1, d1 and d6. All animal experiments were performed under approved IACUC protocols held at the University of Washington and Biogen. Recombinant protein and antibody generation and characterization Human CTGF domain I (sequence Met1 CPDEPAPRCPAGVSLVLDGCGCCRVCAKQLGELCTERDPCDPHKGLFC), domain I+II (sequence Met1CPDEPAPRCPAGVSLVLDGCGCCRVCAKQLGELCTERDPCDPHKGLFCCIFGGT VYRSGESFQSSCKYQCTCLDGAVGCMPLCSMDVRLPSPDCPFPRRVKLPGKCCEE) were cloned and expressed in 293 cells, and purified by Chelating SFF(Ni) Column, tested for single band by SEC and PAGE, and tested for absence of contamination. Domain-IV (sequence GKKCIRTPKISKPIKFELSGCTSMKTYRAKFCGVCTDGRCCTPHRTTTLPVEFKCPDGE VMKKNMMFIKTCACHYNCPGDNDIFESLYYRKMY) was purchased from Peprotech. Mouse or human DKK1 was generated from the coding sequence with some modifications and a tag. Secreted protein was harvested from 293 cells, and purified by nickel column, and tested for activity in a supertopflash (STF) assay 4. DKK1 showed EC50 of 0.69nM for WNT3a-induced WNT signaling in STF cells. -

PRODUCT SPECIFICATION Prest Antigen OCEL1 Product Datasheet
PrEST Antigen OCEL1 Product Datasheet PrEST Antigen PRODUCT SPECIFICATION Product Name PrEST Antigen OCEL1 Product Number APrEST82482 Gene Description occludin/ELL domain containing 1 Alternative Gene FLJ22709 Names Corresponding Anti-OCEL1 (HPA042070) Antibodies Description Recombinant protein fragment of Human OCEL1 Amino Acid Sequence Recombinant Protein Epitope Signature Tag (PrEST) antigen sequence: HKAKTKKIVFEDELLSQALLGAKKPIGAIPKGHKPRPHPVPDYELKYPPV SSERERSRYVAVFQDQYGEFLELQH Fusion Tag N-terminal His6ABP (ABP = Albumin Binding Protein derived from Streptococcal Protein G) Expression Host E. coli Purification IMAC purification Predicted MW 26 kDa including tags Usage Suitable as control in WB and preadsorption assays using indicated corresponding antibodies. Purity >80% by SDS-PAGE and Coomassie blue staining Buffer PBS and 1M Urea, pH 7.4. Unit Size 100 µl Concentration Lot dependent Storage Upon delivery store at -20°C. Avoid repeated freeze/thaw cycles. Notes Gently mix before use. Optimal concentrations and conditions for each application should be determined by the user. Product of Sweden. For research use only. Not intended for pharmaceutical development, diagnostic, therapeutic or any in vivo use. No products from Atlas Antibodies may be resold, modified for resale or used to manufacture commercial products without prior written approval from Atlas Antibodies AB. Warranty: The products supplied by Atlas Antibodies are warranted to meet stated product specifications and to conform to label descriptions when used and stored properly. Unless otherwise stated, this warranty is limited to one year from date of sales for products used, handled and stored according to Atlas Antibodies AB's instructions. Atlas Antibodies AB's sole liability is limited to replacement of the product or refund of the purchase price.