Genomic Approaches to Identify Important Traits in Avian Species Bhuwan Khatri University of Arkansas, Fayetteville
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Implications in Parkinson's Disease
Journal of Clinical Medicine Review Lysosomal Ceramide Metabolism Disorders: Implications in Parkinson’s Disease Silvia Paciotti 1,2 , Elisabetta Albi 3 , Lucilla Parnetti 1 and Tommaso Beccari 3,* 1 Laboratory of Clinical Neurochemistry, Department of Medicine, University of Perugia, Sant’Andrea delle Fratte, 06132 Perugia, Italy; [email protected] (S.P.); [email protected] (L.P.) 2 Section of Physiology and Biochemistry, Department of Experimental Medicine, University of Perugia, Sant’Andrea delle Fratte, 06132 Perugia, Italy 3 Department of Pharmaceutical Sciences, University of Perugia, Via Fabretti, 06123 Perugia, Italy; [email protected] * Correspondence: [email protected] Received: 29 January 2020; Accepted: 20 February 2020; Published: 21 February 2020 Abstract: Ceramides are a family of bioactive lipids belonging to the class of sphingolipids. Sphingolipidoses are a group of inherited genetic diseases characterized by the unmetabolized sphingolipids and the consequent reduction of ceramide pool in lysosomes. Sphingolipidoses include several disorders as Sandhoff disease, Fabry disease, Gaucher disease, metachromatic leukodystrophy, Krabbe disease, Niemann Pick disease, Farber disease, and GM2 gangliosidosis. In sphingolipidosis, lysosomal lipid storage occurs in both the central nervous system and visceral tissues, and central nervous system pathology is a common hallmark for all of them. Parkinson’s disease, the most common neurodegenerative movement disorder, is characterized by the accumulation and aggregation of misfolded α-synuclein that seem associated to some lysosomal disorders, in particular Gaucher disease. This review provides evidence into the role of ceramide metabolism in the pathophysiology of lysosomes, highlighting the more recent findings on its involvement in Parkinson’s disease. Keywords: ceramide metabolism; Parkinson’s disease; α-synuclein; GBA; GLA; HEX A-B; GALC; ASAH1; SMPD1; ARSA * Correspondence [email protected] 1. -

Coupling of Spliceosome Complexity to Intron Diversity
bioRxiv preprint doi: https://doi.org/10.1101/2021.03.19.436190; this version posted March 20, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Coupling of spliceosome complexity to intron diversity Jade Sales-Lee1, Daniela S. Perry1, Bradley A. Bowser2, Jolene K. Diedrich3, Beiduo Rao1, Irene Beusch1, John R. Yates III3, Scott W. Roy4,6, and Hiten D. Madhani1,6,7 1Dept. of Biochemistry and Biophysics University of California – San Francisco San Francisco, CA 94158 2Dept. of Molecular and Cellular Biology University of California - Merced Merced, CA 95343 3Department of Molecular Medicine The Scripps Research Institute, La Jolla, CA 92037 4Dept. of Biology San Francisco State University San Francisco, CA 94132 5Chan-Zuckerberg Biohub San Francisco, CA 94158 6Corresponding authors: [email protected], [email protected] 7Lead Contact 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.03.19.436190; this version posted March 20, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. SUMMARY We determined that over 40 spliceosomal proteins are conserved between many fungal species and humans but were lost during the evolution of S. cerevisiae, an intron-poor yeast with unusually rigid splicing signals. We analyzed null mutations in a subset of these factors, most of which had not been investigated previously, in the intron-rich yeast Cryptococcus neoformans. -

Gene Expression Studies: from Case-Control to Multiple-Population-Based Studies
From the Institute of Human Genetics, Helmholtz Zentrum Munchen,¨ Deutsches Forschungszentrum fur¨ Gesundheit und Umwelt (GmbH) Head: Prof. Dr. Thomas Meitinger Gene expression studies: From case-control to multiple-population-based studies Thesis Submitted for a Doctoral Degree in Natural Sciences at the Faculty of Medicine, Ludwig-Maximilians-Universitat¨ Munchen¨ Katharina Schramm Dachau, Germany 2016 With approval of the Faculty of Medicine Ludwig-Maximilians-Universit¨atM ¨unchen Supervisor/Examiner: Prof. Dr. Thomas Illig Co-Examiners: Prof. Dr. Roland Kappler Dean: Prof. Dr. med. dent. Reinhard Hickel Date of oral examination: 22.12.2016 II Dedicated to my family. III Abstract Recent technological developments allow genome-wide scans of gene expression levels. The reduction of costs and increasing parallelization of processing enable the quantification of 47,000 transcripts in up to twelve samples on a single microarray. Thereby the data collec- tion of large population-based studies was improved. During my PhD, I first developed a workflow for the statistical analyses of case-control stu- dies of up to 50 samples. With large population-based data sets generated I established a pipeline for quality control, data preprocessing and correction for confounders, which re- sulted in substantially improved data. In total, I processed more than 3,000 genome-wide expression profiles using the generated pipeline. With 993 whole blood samples from the population-based KORA (Cooperative Health Research in the Region of Augsburg) study we established one of the largest population-based resource. Using this data set we contributed to a number of transcriptome-wide association studies within national (MetaXpress) and international (CHARGE) consortia. -
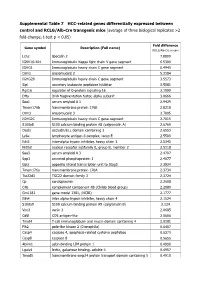
Supplemental Table 7 HCC-Related Genes Differentially Expressed Between Control and RCLG/Alb-Cre Transgenic Mice
Supplemental Table 7 HCC-related genes differentially expressed between control and RCLG/Alb-Cre transgenic mice (average of three biological replicates >2 fold-change, t-test p < 0.05) Fold difference Gene symbol Description (Full name) (RCLG/Alb-Cre vs con) Lcn2 lipocalin 2 7.8899 IGKV16-104 Immunoglobulin Kappa light chain V gene segment 6.5300 IGHG1 Immunoglobulin heavy chain C gene segment 6.4945 Orm2 orosomucoid 2 5.3184 IGHG2B Immunoglobulin heavy chain C gene segment 3.5573 Slpi secretory leukocyte peptidase inhibitor 3.5081 Rgs16 regulator of G-protein signaling 16 3.1999 Dffa DNA fragmentation factor, alpha subunit 3.0666 Saa1 serum amyloid A 1 2.9429 Tmem176b transmembrane protein 176B 2.8218 Orm3 orosomucoid 3 2.7805 IGHG2C Immunoglobulin heavy chain C gene segment 2.7015 S100a8 S100 calcium binding protein A8 (calgranulin A) 2.6769 Ocel1 occludin/ELL domain containing 1 2.6553 Ly6e lymphocyte antigen 6 complex, locus E 2.5509 Itih3 inter-alpha trypsin inhibitor, heavy chain 3 2.5345 Nr0b2 nuclear receptor subfamily 0, group B, member 2 2.5118 Saa3 serum amyloid A 3 2.4707 Spp1 secreted phosphoprotein 1 2.4077 Gats opposite strand transcription unit to Stag3 2.3934 Tmem176a transmembrane protein 176A 2.3734 Tsc22d3 TSC22 domain family 3 2.3724 Cp ceruloplasmin 2.2608 C4b complement component 4B (Childo blood group) 2.2089 Gm1381 gene model 1381, (NCBI) 2.1777 Itih4 inter alpha-trypsin inhibitor, heavy chain 4 2.1524 S100a9 S100 calcium binding protein A9 (calgranulin B) 2.124 Vnn3 vanin 3 2.0685 Cd5l CD5 antigen-like 2.0606 -

4-6 Weeks Old Female C57BL/6 Mice Obtained from Jackson Labs Were Used for Cell Isolation
Methods Mice: 4-6 weeks old female C57BL/6 mice obtained from Jackson labs were used for cell isolation. Female Foxp3-IRES-GFP reporter mice (1), backcrossed to B6/C57 background for 10 generations, were used for the isolation of naïve CD4 and naïve CD8 cells for the RNAseq experiments. The mice were housed in pathogen-free animal facility in the La Jolla Institute for Allergy and Immunology and were used according to protocols approved by the Institutional Animal Care and use Committee. Preparation of cells: Subsets of thymocytes were isolated by cell sorting as previously described (2), after cell surface staining using CD4 (GK1.5), CD8 (53-6.7), CD3ε (145- 2C11), CD24 (M1/69) (all from Biolegend). DP cells: CD4+CD8 int/hi; CD4 SP cells: CD4CD3 hi, CD24 int/lo; CD8 SP cells: CD8 int/hi CD4 CD3 hi, CD24 int/lo (Fig S2). Peripheral subsets were isolated after pooling spleen and lymph nodes. T cells were enriched by negative isolation using Dynabeads (Dynabeads untouched mouse T cells, 11413D, Invitrogen). After surface staining for CD4 (GK1.5), CD8 (53-6.7), CD62L (MEL-14), CD25 (PC61) and CD44 (IM7), naïve CD4+CD62L hiCD25-CD44lo and naïve CD8+CD62L hiCD25-CD44lo were obtained by sorting (BD FACS Aria). Additionally, for the RNAseq experiments, CD4 and CD8 naïve cells were isolated by sorting T cells from the Foxp3- IRES-GFP mice: CD4+CD62LhiCD25–CD44lo GFP(FOXP3)– and CD8+CD62LhiCD25– CD44lo GFP(FOXP3)– (antibodies were from Biolegend). In some cases, naïve CD4 cells were cultured in vitro under Th1 or Th2 polarizing conditions (3, 4). -

Supporting Information
Supporting Information Muro et al. 10.1073/pnas.0807813105 SI Text (Fig. S1) showed for 10 of the PAS variants a marked tendency 1. Algorithm for Automated Recognition of EST End Clusters. To to lie 10–30 nt upstream of EST ends, whereas no strong trend determine the coordinates of genomic EST alignments we used was seen for 3 additional variants. Based on this simple analysis the UCSC genome annotation (1), a regularly updated database we chose to accept only 10 of the variants as functional PAS which includes mouse and human EST sequences and their (Table S1), and only EST ends validated by a PAS 10–30 nt positions in the genome. The UCSC Golden Path version was upstream. Furthermore, we considered all EST ends validated by mm6 for mouse (equivalent to NCBI build 34, March 2005) and the same PAS and ending within 20 nt of one another to hg18 for human (NCBI Build 36.1, March 2006). The UCSC represent the same transcript end and to be clustered together, mapping database is based on alignments of ESTs to the as others have also proposed (6). corresponding genome using the BLAT program (2). Because of Next, we studied the distribution of individual EST ends the relatively low accuracy of EST sequencing and recent around our predicted rough ends (Fig. S2). The graph indicates genomic duplications (3), some ESTs can be aligned to more that in a range of Ϫ200 to ϩ150 nt around these rough ends the than 1 genomic position. To avoid possible misidentifications, frequency of EST ends is distinguishable from the surrounding ambiguously aligning ESTs were excluded from our analysis. -

S41467-020-18249-3.Pdf
ARTICLE https://doi.org/10.1038/s41467-020-18249-3 OPEN Pharmacologically reversible zonation-dependent endothelial cell transcriptomic changes with neurodegenerative disease associations in the aged brain Lei Zhao1,2,17, Zhongqi Li 1,2,17, Joaquim S. L. Vong2,3,17, Xinyi Chen1,2, Hei-Ming Lai1,2,4,5,6, Leo Y. C. Yan1,2, Junzhe Huang1,2, Samuel K. H. Sy1,2,7, Xiaoyu Tian 8, Yu Huang 8, Ho Yin Edwin Chan5,9, Hon-Cheong So6,8, ✉ ✉ Wai-Lung Ng 10, Yamei Tang11, Wei-Jye Lin12,13, Vincent C. T. Mok1,5,6,14,15 &HoKo 1,2,4,5,6,8,14,16 1234567890():,; The molecular signatures of cells in the brain have been revealed in unprecedented detail, yet the ageing-associated genome-wide expression changes that may contribute to neurovas- cular dysfunction in neurodegenerative diseases remain elusive. Here, we report zonation- dependent transcriptomic changes in aged mouse brain endothelial cells (ECs), which pro- minently implicate altered immune/cytokine signaling in ECs of all vascular segments, and functional changes impacting the blood–brain barrier (BBB) and glucose/energy metabolism especially in capillary ECs (capECs). An overrepresentation of Alzheimer disease (AD) GWAS genes is evident among the human orthologs of the differentially expressed genes of aged capECs, while comparative analysis revealed a subset of concordantly downregulated, functionally important genes in human AD brains. Treatment with exenatide, a glucagon-like peptide-1 receptor agonist, strongly reverses aged mouse brain EC transcriptomic changes and BBB leakage, with associated attenuation of microglial priming. We thus revealed tran- scriptomic alterations underlying brain EC ageing that are complex yet pharmacologically reversible. -

Investigation of the Underlying Hub Genes and Molexular Pathogensis in Gastric Cancer by Integrated Bioinformatic Analyses
bioRxiv preprint doi: https://doi.org/10.1101/2020.12.20.423656; this version posted December 22, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Investigation of the underlying hub genes and molexular pathogensis in gastric cancer by integrated bioinformatic analyses Basavaraj Vastrad1, Chanabasayya Vastrad*2 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karanataka, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India bioRxiv preprint doi: https://doi.org/10.1101/2020.12.20.423656; this version posted December 22, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract The high mortality rate of gastric cancer (GC) is in part due to the absence of initial disclosure of its biomarkers. The recognition of important genes associated in GC is therefore recommended to advance clinical prognosis, diagnosis and and treatment outcomes. The current investigation used the microarray dataset GSE113255 RNA seq data from the Gene Expression Omnibus database to diagnose differentially expressed genes (DEGs). Pathway and gene ontology enrichment analyses were performed, and a proteinprotein interaction network, modules, target genes - miRNA regulatory network and target genes - TF regulatory network were constructed and analyzed. Finally, validation of hub genes was performed. The 1008 DEGs identified consisted of 505 up regulated genes and 503 down regulated genes. -

Common Variant Near the Endothelin Receptor Type a (EDNRA)
Common variant near the endothelin receptor type A(EDNRA) gene is associated with intracranial aneurysm risk Katsuhito Yasunoa,b,c, Mehmet Bakırcıog˘ lua,b,c, Siew-Kee Lowd, Kaya Bilgüvara,b,c, Emília Gaála,b,c,e, Ynte M. Ruigrokf, Mika Niemeläe, Akira Hatag, Philippe Bijlengah, Hidetoshi Kasuyai, Juha E. Jääskeläinenj, Dietmar Krexk, Georg Auburgerl, Matthias Simonm, Boris Krischekn, Ali K. Ozturka,b,c, Shrikant Maneo, Gabriel J. E. Rinkelf, Helmuth Steinmetzl, Juha Hernesniemie, Karl Schallerh, Hitoshi Zembutsud, Ituro Inouep, Aarno Palotieq, François Cambienr, Yusuke Nakamurad, Richard P. Liftonc,s,t,1, and Murat Günela,b,c,1 Departments of aNeurosurgery, bNeurobiology, and cGenetics, Yale Program on Neurogenetics, Yale Center for Human Genetics and Genomics, and sDepartment of Internal Medicine and tHoward Hughes Medical Institute, Yale University School of Medicine, New Haven, CT 06510; dHuman Genome Center, Institute of Medical Science, University of Tokyo, Tokyo 108-8639, Japan; eDepartment of Neurosurgery, Helsinki University Central Hospital, Helsinki, FI-00029 HUS, Finland; fDepartment of Neurology, Rudolf Magnus Institute of Neuroscience, University Medical Center Utrecht, 3584 CX Utrecht, The Netherlands; gDepartment of Public Health, School of Medicine, Chiba University, Chiba 260-8670, Japan; hService de Neurochirurgie, Department of Clinical Neurosciences, Geneva University Hospital, 1211 Geneva 4, Switzerland; iDepartment of Neurosurgery, Medical Center East, Tokyo Women’s University, Tokyo 116-8567, Japan; jDepartment -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

RRBP1 (NM 001042576) Human Tagged ORF Clone Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for RC218816L3 RRBP1 (NM_001042576) Human Tagged ORF Clone Product data: Product Type: Expression Plasmids Product Name: RRBP1 (NM_001042576) Human Tagged ORF Clone Tag: Myc-DDK Symbol: RRBP1 Synonyms: ES/130; ES130; hES; p180; RRp Vector: pLenti-C-Myc-DDK-P2A-Puro (PS100092) E. coli Selection: Chloramphenicol (34 ug/mL) Cell Selection: Puromycin ORF Nucleotide The ORF insert of this clone is exactly the same as(RC218816). Sequence: Restriction Sites: SgfI-MluI Cloning Scheme: ACCN: NM_001042576 ORF Size: 2931 bp This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 2 RRBP1 (NM_001042576) Human Tagged ORF Clone – RC218816L3 OTI Disclaimer: Due to the inherent nature of this plasmid, standard methods to replicate additional amounts of DNA in E. coli are highly likely to result in mutations and/or rearrangements. Therefore, OriGene does not guarantee the capability to replicate this plasmid DNA. Additional amounts of DNA can be purchased from OriGene with batch-specific, full-sequence verification at a reduced cost. Please contact our customer care team at [email protected] or by calling 301.340.3188 option 3 for pricing and delivery. The molecular sequence of this clone aligns with the gene accession number as a point of reference only. However, individual transcript sequences of the same gene can differ through naturally occurring variations (e.g. -

Loss of Conserved Ubiquitylation Sites in Conserved Proteins During Human Evolution
INTERNATIONAL JOURNAL OF MOleCular meDICine 42: 2203-2212, 2018 Loss of conserved ubiquitylation sites in conserved proteins during human evolution DONGBIN PARK, CHUL JUN GOH, HYEIN KIM, JI SEOK LEE and YOONSOO HAHN Department of Life Science, Chung‑Ang University, Seoul 06974, Republic of Korea Received January 30, 2018; Accepted July 6, 2018 DOI: 10.3892/ijmm.2018.3772 Abstract. Ubiquitylation of lysine residues in proteins serves Introduction a pivotal role in the efficient removal of misfolded or unused proteins and in the control of various regulatory pathways Ubiquitylation, in which the highly conserved 76‑residue poly- by monitoring protein activity that may lead to protein peptide ubiquitin is covalently attached to a lysine residue of degradation. The loss of ubiquitylated lysines may affect substrate proteins, mediates the targeted destruction of ubiq- the ubiquitin‑mediated regulatory network and result in the uitylated proteins by the ubiquitin‑proteasome system (1‑4). emergence of novel phenotypes. The present study analyzed The ubiquitin‑mediated protein degradation pathway serves a mouse ubiquitylation data and orthologous proteins from crucial role in the efficient and specific removal of misfolded 62 mammals to identify 193 conserved ubiquitylation sites from proteins and certain key regulatory proteins (5,6). Ubiquitin 169 proteins that were lost in the Euarchonta lineage leading and other ubiquitin‑like proteins, including autophagy‑related to humans. A total of 8 proteins, including betaine homo- protein 8, Ubiquitin‑like