The Structural Basis of ATP As an Allosteric Modulator
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

| Hai Lui a Un Acutul Luniit Moonhiti
|HAI LUI AUN ACUTULUS010006055B2 LUNIIT MOONHITI (12 ) United States Patent (10 ) Patent No. : US 10 , 006 , 055 B2 Burk et al. (45 ) Date of Patent: Jun . 26 , 2018 ( 54 ) MICROORGANISMS FOR PRODUCING 2002/ 0168654 A1 11/ 2002 Maranas et al. 2003 / 0059792 Al 3 /2003 Palsson et al . BUTADIENE AND METHODS RELATED 2003 /0087381 A1 5 / 2003 Gokarn THERETO 2003 / 0224363 Al 12 /2003 Park et al . 2003 / 0233218 Al 12 /2003 Schilling (71 ) Applicant: Genomatica , Inc. , San Diego , CA (US ) 2004 / 0009466 AL 1 /2004 Maranas et al. 2004 / 0029149 Al 2 /2004 Palsson et al. ( 72 ) Inventors : Mark J . Burk , San Diego , CA (US ) ; 2004 / 0072723 A1 4 /2004 Palsson et al. Anthony P . Burgard , Bellefonte , PA 2004 / 0152159 Al 8 / 2004 Causey et al . 2005 /0042736 A1 2 / 2005 San et al . (US ) ; Robin E . Osterhout , San Diego , 2005 / 0079482 A1 4 / 2005 Maranas et al . CA (US ) ; Jun Sun , San Diego , CA 2006 / 0046288 Al 3 / 2006 Ka - Yiu et al. ( US ) ; Priti Pharkya , San Diego , CA 2006 / 0073577 A1 4 / 2006 Ka - Yiu et al . (US ) 2007 /0184539 Al 8 / 2007 San et al . 2009 / 0047718 Al 2 / 2009 Blaschek et al . 2009 / 0047719 Al 2 / 2009 Burgard et al . (73 ) Assignee : Genomatica , Inc ., San Diego , CA (US ) 2009 /0191593 A1 7 / 2009 Burk et al . 2010 / 0003716 A1 1 / 2010 Cervin et al. ( * ) Notice : Subject to any disclaimer , the term of this 2010 /0184171 Al 7 /2010 Jantama et al. patent is extended or adjusted under 35 2010 /0304453 Al 12 / 2010 Trawick et al . -

(12) Patent Application Publication (10) Pub. No.: US 2014/0155567 A1 Burk Et Al
US 2014O155567A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2014/0155567 A1 Burk et al. (43) Pub. Date: Jun. 5, 2014 (54) MICROORGANISMS AND METHODS FOR (60) Provisional application No. 61/331,812, filed on May THE BIOSYNTHESIS OF BUTADENE 5, 2010. (71) Applicant: Genomatica, Inc., San Diego, CA (US) Publication Classification (72) Inventors: Mark J. Burk, San Diego, CA (US); (51) Int. Cl. Anthony P. Burgard, Bellefonte, PA CI2P 5/02 (2006.01) (US); Jun Sun, San Diego, CA (US); CSF 36/06 (2006.01) Robin E. Osterhout, San Diego, CA CD7C II/6 (2006.01) (US); Priti Pharkya, San Diego, CA (52) U.S. Cl. (US) CPC ................. CI2P5/026 (2013.01); C07C II/I6 (2013.01); C08F 136/06 (2013.01) (73) Assignee: Genomatica, Inc., San Diego, CA (US) USPC ... 526/335; 435/252.3:435/167; 435/254.2: (21) Appl. No.: 14/059,131 435/254.11: 435/252.33: 435/254.21:585/16 (22) Filed: Oct. 21, 2013 (57) ABSTRACT O O The invention provides non-naturally occurring microbial Related U.S. Application Data organisms having a butadiene pathway. The invention addi (63) Continuation of application No. 13/101,046, filed on tionally provides methods of using Such organisms to produce May 4, 2011, now Pat. No. 8,580,543. butadiene. Patent Application Publication Jun. 5, 2014 Sheet 1 of 4 US 2014/O155567 A1 ?ueudos!SMS |?un61– Patent Application Publication Jun. 5, 2014 Sheet 2 of 4 US 2014/O155567 A1 VOJ OO O Z?un61– Patent Application Publication US 2014/O155567 A1 {}}} Hººso Patent Application Publication Jun. -
Generate Metabolic Map Poster
Authors: Peter D. Karp Suzanne Paley Julio Collado-Vides John L Ingraham Ingrid Keseler Markus Krummenacker Cesar Bonavides-Martinez Robert Gunsalus An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Carol Fulcher Ian Paulsen Socorro Gama-Castro Robert LaRossa Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. EcoCyc: Escherichia coli K-12 substr. MG1655 Cellular Overview Connections between pathways are omitted for legibility. Anamika Kothari Amanda Mackie Alberto Santos-Zavaleta succinate phosphate succinate N-acetyl-DL-methionine + L-ornithine glutathione + L-methionine S-oxide D-fructofuranose γ Ag+ molybdate ferroheme b L,L-homocystine asp lys cys L-alanyl- -D- D-mannopyranose 6-phosphate 2+ 2+ H D-methionine 2-deoxy-D-glucose succinate formate formate succinate D-tartrate putrescine agmatine cadaverine L-tartrate D-fructofuranose 6-phosphate + nitrate nitrate Cu thiosulfate deoxycholate L,L-homocystine D-cystine D-cycloserine methyl β-D-glucoside putrescine asp spermidine (S)-2-hydroxybutanoate (S)-2-hydroxybutanoate arg L-homoserine lactone magnesium hydrogenphosphate magnesium hydrogenphosphate antimonous acid glutamyl-meso- Co2+ Cd2+ lactulose poly-β-1,6- met cob(I)inamide 2,3-dioxo- -

Characterization of Human UMP/CMP Kinase and Its Phosphorylation of D- and 1 L-Form Deoxycytidine Analogue Monophosphates
[CANCER RESEARCH 62, 1624–1631, March 15, 2002] Characterization of Human UMP/CMP Kinase and Its Phosphorylation of D- and 1 L-Form Deoxycytidine Analogue Monophosphates Jieh-Yuan Liou, Ginger E. Dutschman, Wing Lam, Zaoli Jiang, and Yung-Chi Cheng2 Department of Pharmacology, Yale University School of Medicine, New Haven, Connecticut 06520 ABSTRACT with leukemia, lymphoma, or solid tumors (11). Deoxycytidine ana- logues, such as -D-2Ј,3Ј-dideoxycytidine and L-(Ϫ)-SddC (Lamivu- Pyrimidine nucleoside monophosphate kinase [UMP/CMP kinase dine), have been shown to have anti-HIV and antihuman hepatitis B (UMP/CMPK); EC 2.7.4.14] plays a crucial role in the formation of UDP, virus activities (12–17). L-(Ϫ)-SddC was the first nucleoside analogue CDP, and dCDP, which are required for cellular nucleic acid synthesis. Several cytidine and deoxycytidine analogues are important anticancer with an L configuration to show therapeutic activity and, thus, defined  Ј Ј and antiviral drugs. These drugs require stepwise phosphorylation to their a new category for the design of nucleoside analogues. -L-2 ,3 - triphosphate forms to exert their therapeutic effects. The role of UMP/ dideoxy-5-fluoro-3Ј-thia-cytidine and -L-2Ј,3Ј-dideoxy-2Ј,3Ј-dide- CMPK for the phosphorylation of nucleoside analogues has been indi- hydro-5-fluorocytidine have been shown to be potent antihuman hep- cated. Thus, we cloned the human UMP/CMPK gene, expressed it in atitis B virus agents in vitro and in animal studies (18–22). In studies Escherichia coli, and purified it to homogeneity. Its kinetic properties of other -L-(Ϫ)-2Ј,3Ј-dideoxycytidine analogues, it was observed were determined. -

Saccharomyces Cerevisiae Aspartate Kinase Mechanism and Inhibition
In compliance with the Canadian Privacy Legislation some supporting forms may have been removed from this dissertation. While these forms may be included in the document page count, their removal does not represent any loss of content from the dissertation. Ph.D. Thesis - D. Bareich McMaster University - Department of Biochemistry FUNGAL ASPARTATE KINASE MECHANISM AND INHIBITION By DAVID C. BAREICH, B.Sc. A Thesis Submitted to the School of Graduate Studies in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy McMaster University © Copyright by David C. Bareich, June 2003 1 Ph.D. Thesis - D. Bareich McMaster University - Department of Biochemistry FUNGAL ASPARTATE KINASE MECHANISM AND INHIBITION Ph.D. Thesis - D. Bareich McMaster University - Department of Biochemistry DOCTOR OF PHILOSOPHY (2003) McMaster University (Biochemistry) Hamilton, Ontario TITLE: Saccharomyces cerevisiae aspartate kinase mechanism and inhibition AUTHOR: David Christopher Bareich B.Sc. (University of Waterloo) SUPERVISOR: Professor Gerard D. Wright NUMBER OF PAGES: xix, 181 11 Ph.D. Thesis - D. Bareich McMaster University - Department of Biochemistry ABSTRACT Aspartate kinase (AK) from Saccharomyces cerevisiae (AKsc) catalyzes the first step in the aspartate pathway responsible for biosynthesis of L-threonine, L-isoleucine, and L-methionine in fungi. Little was known about amino acids important for AKsc substrate binding and catalysis. Hypotheses about important amino acids were tested using site directed mutagenesis to substitute these amino acids with others having different properties. Steady state kinetic parameters and pH titrations of the variant enzymes showed AKsc-K18 and H292 to be important for binding and catalysis. Little was known about how the S. cerevisiae aspartate pathway kinases, AKsc and homoserine kinase (HSKsc), catalyze the transfer of the y-phosphate from adenosine triphosphate (ATP) to L-aspartate or L-homoserine, respectively. -

A NOVEL TWO-DOMAIN ARCHITECTURE WITHIN the AMINO ACID KINASE ENZYME FAMILY REVEALED by the CRYSTAL STRUCTURE of Escherichia Coli
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Digital.CSIC A NOVEL TWO-DOMAIN ARCHITECTURE WITHIN THE AMINO ACID KINASE ENZYME FAMILY REVEALED BY THE CRYSTAL STRUCTURE OF Escherichia coli GLUTAMATE 5-KINASE Clara Marco-Marín1, Fernando Gil-Ortiz,1 Isabel Pérez-Arellano,2 Javier Cervera,2 Ignacio Fita3 and Vicente Rubio1,* 1Instituto de Biomedicina de Valencia (IBV-CSIC) and Center for Biomedical Research on Rare Diseases (CIBERER-ISCIII), Jaume Roig 11, Valencia-46010, Spain 2Centro de Investigación Príncipe Felipe (FVIB-CSIC), Avda. Autopista del Saler 16, Valencia-46013, Spain 3Instituto de Biología Molecular de Barcelona (IBMB-CSIC). Institute for Research in Biomedicine. Parc Científic, Josep Samitier 1-5, 08028-Barcelona, Spain. Present address: F. Gil-Ortiz, Centro de Investigación Príncipe Felipe (FVIB-CSIC), Avda. Autopista del Saler 16, Valencia-46013, Spain * Corresponding author: Vicente Rubio Instituto de Biomedicina de Valencia Jaume Roig 11, Valencia-46010, Spain E-mail: [email protected] Tel. +34 963 391 772 Fax. +34 963 690 800 Short title: Structure of -glutamyl kinase of Escherichia coli 1 Summary. Glutamate 5-kinase (G5K) makes the highly unstable product glutamyl-5- phosphate (G5P) in the initial, controlling step of proline/ornithine synthesis, being feed-back inhibited by proline or ornithine, and causing, when defective, clinical hyperammonaemia. We have determined two crystal structures of G5K from Escherichia coli, at 2.9- and 2.5-Å-resolution, complexed with glutamate and sulphate, or with G5P, sulphate and the proline analog 5-oxoproline. E. coli G5K presents a novel tetrameric (dimer of dimers) architecture. -
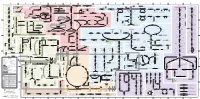
Fullsubwaymap221.Pdf
A B C D E F G H I J K L M Aldose reductase Sorbitol dehydrogenase Cholesterol synthesis Glycogen and galactose metabolism (branch point only) (debranching) glucose sorbitol fructose Aromatic amino acid metabolism Purine synthesis Pyrimidine synthesis glycogen (n+1) phenylacetate triiodothyronine (T3) dopaquinone melanin + + thyroxine (T4) (multiple steps) (many cycles) P 4' NADPH NADP NAD NADH acetyl-CoA x2 i Debranching enzyme 3' - α-1,4 bonds Aromatic amino acid 5-phosphoribosyl pyrophosphate (PRPP) HCO B 2' P 3 (branching) Glycogen 6 i (multiple steps) O H O (multiple steps) Tyrosinase O H O 1' 2 2 2 2 B 1 2 3 4 5 6 7 8 9 10 Pentose phosphate pathway Phenylalanine Tyrosine decarboxylase 6 Dopamine B Thiolase phosphorylase -5 -4 -3 -2 -1 0 1 2 3 4 Transaminase 6 glutamine 2 ATP (many cycles) Glycogen Glucose-6-phosphatase Dehydrogenase hydroxylase hydroxylase (DOPA decarboxylase) β-hydroxylase Methyltransferase 10 UDP 4:4 Transferase ATP glutathione (reduced) H O phenyllactate phenylpyruvate phenylalanine tyrosine dihydroxy- dopamine Glutamine PRPP CoA synthase Hexokinase or (in endoplasmic reticulum) 2 2 norepinephrine epinephrine glutamine Carbamoyl 9 1' phenylanine amidotransferase (GPAT) glutamate glycogen (n) Glutathione reductase Glutathione peroxidase + phosphate glycogen (n) -5 -4 -3 -2 -1 0 1 2 3 4 2' 3' 4' glucokinase 8 NADP NADPH glutamate α-ketoglutarate CO2 O H O Glycosyl (4:6) UDP-glucose ADP (L-DOPA) 2 2 synthetase II glutathione (oxidized) 2 H O α-ketoglutarate PPi glutamate acetoacetyl-CoA 7 1 Glucose -1,6-Glucosidase -

Generated by SRI International Pathway Tools Version 25.0, Authors S
Authors: Pallavi Subhraveti Ron Caspi Peter Midford Peter D Karp An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_000190595Cyc: Cellulophaga lytica DSM 7489 Cellular Overview Connections between pathways are omitted for legibility. Anamika Kothari phosphate molybdate RS04350 RS08315 phosphate molybdate Macromolecule Modification tRNA-uridine 2-thiolation inosine 5'- Nucleoside and Nucleotide Degradation ditrans,octacis- a peptidoglycan with ditrans,octacis- a [bis(guanylyl an N-terminal- a [protein] and selenation (bacteria) phosphate guanosine ditrans,octacis- (4R)-4-hydroxy- an L-asparaginyl- an L-cysteinyl- Aminoacyl-tRNA Charging tRNA charging nucleotides undecaprenyldiphospho- (L-alanyl-γ-D-glutamyl- undecaprenyldiphospho- undecaprenyldiphospho- molybdopterin) cys L-methionyl-L- C-terminal glu degradation 2-oxoglutarate [tRNA Asn ] [tRNA Cys ] degradation III N-acetyl-(N-acetyl-β-D- L-lysyl-D-alanyl-D- N-acetyl-(N- N-acetyl-(N- cofactor cysteinyl-[protein] L-glutamate cys glucosaminyl)muramoyl- alanine) pentapeptide acetylglucosaminyl) acetylglucosaminyl) chaperone]- Phe IMP bifunctional phe a tRNA L-alanyl-γ-D-glutamyl-L- -

Exchange of Nucleoside Monophosphate-Binding Domains in Adenylate Kinase and UMP/CMP Kinase1
J. Biochem. 124, 359-367 (1998) Exchange of Nucleoside Monophosphate-Binding Domains in Adenylate Kinase and UMP/CMP Kinase1 Toshihide Okajima,*"z Tamo Fukamizo,* Sachio Goto,* Toshio Fukui,t and Katsuyuki Tanizawa•õ *Faculty of Agriculture , Kinki University, 3327-204 Nakamachi, Nara 631-8505; and •õInstitute of Scientific and Industrial Research, Osaka University, Ibaraki, Osaka 567-0047 Received for publication, March 6, 1998 Two types of active chimeric enzymes have been constructed by genetic engineering of chicken cytosolic adenylate kinase (AK) and porcine brain UMP/CMP kinase (UCK): one, designated as UAU, carries an AMP-binding domain of AK in the remaining body of UCK; and the other, designated as AUA, carries a UMP/CMP-binding domain of UCK in the remaining body of AK. Steady-state kinetic analysis of these chimeric enzymes revealed that UAU is 4-fold more active for AMP, 40-fold less active for UMP, and 4-fold less active for CMP than the parental UCK, although AUA has considerably lowered reactivity for both AMP and UMP. Circular dichroism spectra of the two chimeric enzymes suggest that UAU and AUA have similar folding structures to UCK and AK, respectively. Furthermore, proton NMR measurements of the UCK and UAU proteins indicate that significant differ ences in proton signals are limited to the aromatic region, where an imidazole C2H signal assigned to His31 shows a downfield shift upon conversion of UCK to UAU, and the signals assigned to Tyr49 and Tyr56 in the UMP/CMP-binding domain disappear in UAU. In contrast, AUA has a Tm value about 11•Ž lower than AK, whereas UAU and UCK have similar Tm values. -

Complex Bacterial Consortia Reprogram the Colitogenic Activity of Enterococcus Faecalis in a Gnotobiotic Mouse Model of Chronic Immune-Mediated Colitis
TECHNISCHE UNIVERSITÄT MÜNCHEN Lehrstuhl für Ernährung und Immunologie Complex bacterial consortia reprogram the colitogenic activity of Enterococcus faecalis in a gnotobiotic mouse model of chronic immune-mediated colitis Isabella Brigitta Lengfelder Vollständiger Abdruck der von der Fakultät Wissenschaftszentrum Weihenstephan für Ernährung, Landnutzung und Umwelt der Technischen Universität München zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften (Dr. rer. nat.) genehmigten Dissertation. Vorsitzender: Prof. Dr. Siegfried Scherer Prüfer der Dissertation: 1. Prof. Dr. Dirk Haller 2. Prof. Dr. Wolfgang Liebl Die Dissertation wurde am 01.07.2019 bei der Technischen Universität München eingereicht und durch die Fakultät Wissenschaftszentrum Weihenstephan für Ernährung, Landnutzung und Umwelt am 11.10.2019 angenommen. I ABSTRACT ABSTRACT Inflammatory bowel diseases (IBD) are associated with compositional and functional changes of the intestinal microbiota, but specific contributions of individual bacteria to chronic intestinal inflammation remain unclear. Enterococcus faecalis is a resident member of the human intestinal core microbiota that has been linked to the pathogenesis of IBD and induces chronic colitis in susceptible monoassociated IL10-deficient (IL-10-/-) mice. In this study, we characterized the colitogenic activity of E. faecalis as part of a simplified microbial consortium based on human strains (SIHUMI). RNA sequencing analysis of E. faecalis isolated from monoassociated wild type and IL-10-/- mice identified 408 genes including 14 genes of the ethanolamine utilization (eut) locus to be significantly upregulated in response to inflammation. Despite considerable upregulation of eut genes, deletion of ethanolamine utilization (∆eutVW) had no impact on E. faecalis colitogenic activity in monoassociated IL-10-/- mice. However, replacement of the E. faecalis wild type bacteria by a ∆eutVW mutant in SIHUMI-colonized IL-10-/- mice resulted in exacerbated colitis, suggesting protective functions of E. -
Showdomycin, a New Nucleoside Antibiotic'
[CANCER RESEARCH 28, 1106-1610,Aug@at1968] Showdomycin, A New Nucleoside Antibiotic' S. Roy-Burman,P.Roy-Burman,and D. W. Visser Department of Biochemistry, University of Southern California, School of Medii*ie, Los Angeles, California 9008$ SUMMARY Streptococcus pyogenes (14). Showdomycin has antitumor activity against Ehrlich ascites cells both in vitro and in vivo The metabolism and inhibitory effects of the carbon-linked and is active against cultured HeLa cells ( 11, 14, 15) . Its nucleoside antibiotic showdomycin were investigated in cell structure has been established as 3-$-o-ribofuranosylmaleimide free preparatiozis of Ehrlich ascites cells. The antibiotic is not (5), a carbon-linked nucleoside antibiotic. a substrate for nucleoside kinase or nucleoside phosphorylase. The structural similarity of showdomycin, uridine, and Showdomycin shows inhibitory effects on certain enzymes in an pseudouridine are apparent from the formulae (Chart 1). Ehrlich ascites cell preparation, which are involved in uridine Darnall et at. (5) have pointed out that showdomycin may be and orotic acid metabolism. It inhibits üridine-5'-monophos viewed as pseudouridine which has lost an -NH group in the phokinase, uridine phosphorylase, and possibly orotidylic acid contraction to a five-membered ring. pyrophosphorylase reactions, but has no effect on the activity The unique structure of showdomycin, its structural simi of uridine kinase or adenosine phosphorylase. Showdomycin larity to uridine, and its biologic properties prompted this in strongly inhibits bovine liver uridine-5'[email protected] vestigation of its metabolism and its effect on various enzymes, dehydrogenase but has no effect on rabbit muscle lactic acid particularly those involved in uridine metabolism. -

Springer Handbook of Enzymes
Dietmar Schomburg Ida Schomburg (Eds.) Springer Handbook of Enzymes Alphabetical Name Index 1 23 © Springer-Verlag Berlin Heidelberg New York 2010 This work is subject to copyright. All rights reserved, whether in whole or part of the material con- cerned, specifically the right of translation, printing and reprinting, reproduction and storage in data- bases. The publisher cannot assume any legal responsibility for given data. Commercial distribution is only permitted with the publishers written consent. Springer Handbook of Enzymes, Vols. 1–39 + Supplements 1–7, Name Index 2.4.1.60 abequosyltransferase, Vol. 31, p. 468 2.7.1.157 N-acetylgalactosamine kinase, Vol. S2, p. 268 4.2.3.18 abietadiene synthase, Vol. S7,p.276 3.1.6.12 N-acetylgalactosamine-4-sulfatase, Vol. 11, p. 300 1.14.13.93 (+)-abscisic acid 8’-hydroxylase, Vol. S1, p. 602 3.1.6.4 N-acetylgalactosamine-6-sulfatase, Vol. 11, p. 267 1.2.3.14 abscisic-aldehyde oxidase, Vol. S1, p. 176 3.2.1.49 a-N-acetylgalactosaminidase, Vol. 13,p.10 1.2.1.10 acetaldehyde dehydrogenase (acetylating), Vol. 20, 3.2.1.53 b-N-acetylgalactosaminidase, Vol. 13,p.91 p. 115 2.4.99.3 a-N-acetylgalactosaminide a-2,6-sialyltransferase, 3.5.1.63 4-acetamidobutyrate deacetylase, Vol. 14,p.528 Vol. 33,p.335 3.5.1.51 4-acetamidobutyryl-CoA deacetylase, Vol. 14, 2.4.1.147 acetylgalactosaminyl-O-glycosyl-glycoprotein b- p. 482 1,3-N-acetylglucosaminyltransferase, Vol. 32, 3.5.1.29 2-(acetamidomethylene)succinate hydrolase, p. 287 Vol.