Transcriptome and Glycome Profiling of Human Hematopoietic Cd133+ and Cd34+ Cells
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Seq2pathway Vignette
seq2pathway Vignette Bin Wang, Xinan Holly Yang, Arjun Kinstlick May 19, 2021 Contents 1 Abstract 1 2 Package Installation 2 3 runseq2pathway 2 4 Two main functions 3 4.1 seq2gene . .3 4.1.1 seq2gene flowchart . .3 4.1.2 runseq2gene inputs/parameters . .5 4.1.3 runseq2gene outputs . .8 4.2 gene2pathway . 10 4.2.1 gene2pathway flowchart . 11 4.2.2 gene2pathway test inputs/parameters . 11 4.2.3 gene2pathway test outputs . 12 5 Examples 13 5.1 ChIP-seq data analysis . 13 5.1.1 Map ChIP-seq enriched peaks to genes using runseq2gene .................... 13 5.1.2 Discover enriched GO terms using gene2pathway_test with gene scores . 15 5.1.3 Discover enriched GO terms using Fisher's Exact test without gene scores . 17 5.1.4 Add description for genes . 20 5.2 RNA-seq data analysis . 20 6 R environment session 23 1 Abstract Seq2pathway is a novel computational tool to analyze functional gene-sets (including signaling pathways) using variable next-generation sequencing data[1]. Integral to this tool are the \seq2gene" and \gene2pathway" components in series that infer a quantitative pathway-level profile for each sample. The seq2gene function assigns phenotype-associated significance of genomic regions to gene-level scores, where the significance could be p-values of SNPs or point mutations, protein-binding affinity, or transcriptional expression level. The seq2gene function has the feasibility to assign non-exon regions to a range of neighboring genes besides the nearest one, thus facilitating the study of functional non-coding elements[2]. Then the gene2pathway summarizes gene-level measurements to pathway-level scores, comparing the quantity of significance for gene members within a pathway with those outside a pathway. -

Supplementary Table 2 Gene Sets Used in GSEA
Supplementary Table 2 Gene sets used in GSEA Up in RNAi and Sign Confirmed in Inducible Gene Probe Set ID Accession Symbol Gene Title 200660_at NM_005620 S100A11 S100 calcium binding protein A11 (calgizzarin) 200785_s_at NM_002332 LRP1 low density lipoprotein-related protein 1 (alpha-2-macroglobulin receptor) 201325_s_at NM_001423 EMP1 epithelial membrane protein 1 201373_at NM_000445 PLEC1 plectin 1, intermediate filament binding protein 500kDa 201466_s_at NM_002228 JUN v-jun sarcoma virus 17 oncogene homolog (avian) 201952_at AA156721 ALCAM activated leukocyte cell adhesion molecule 202042_at NM_002109 HARS histidyl-tRNA synthetase 202074_s_at NM_021980 OPTN optineurin 202087_s_at NM_001912 CTSL cathepsin L 202588_at NM_000476 AK1 adenylate kinase 1 202609_at NM_004447 EPS8 epidermal growth factor receptor pathway substrate 8 202733_at NM_004199 P4HA2 procollagen-proline, 2-oxoglutarate 4-dioxygenase (proline 4-hydroxylase), alpha polypeptide II 202756_s_at NM_002081 GPC1 glypican 1 202786_at NM_013233 STK39 serine threonine kinase 39 (STE20/SPS1 homolog, yeast) 202859_x_at NM_000584 IL8 interleukin 8 203083_at NM_003247 THBS2 thrombospondin 2 203186_s_at NM_002961 S100A4 S100 calcium binding protein A4 (calcium protein, calvasculin, metastasin, murine placental homolog) 203232_s_at NM_000332 ATXN1 ataxin 1 203233_at NM_000418 IL4R interleukin 4 receptor 203771_s_at AA740186 BLVRA biliverdin reductase A 203821_at NM_001945 HBEGF heparin-binding EGF-like growth factor 203939_at NM_002526 NT5E 5'-nucleotidase, ecto (CD73) 203955_at NM_014811 -
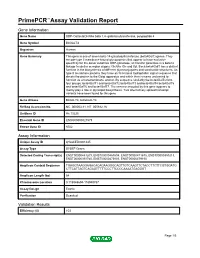
Download Validation Data
PrimePCR™Assay Validation Report Gene Information Gene Name UDP-Gal:betaGlcNAc beta 1,4- galactosyltransferase, polypeptide 4 Gene Symbol B4GALT4 Organism Human Gene Summary This gene is one of seven beta-14-galactosyltransferase (beta4GalT) genes. They encode type II membrane-bound glycoproteins that appear to have exclusive specificity for the donor substrate UDP-galactose; all transfer galactose in a beta14 linkage to similar acceptor sugars: GlcNAc Glc and Xyl. Each beta4GalT has a distinct function in the biosynthesis of different glycoconjugates and saccharide structures. As type II membrane proteins they have an N-terminal hydrophobic signal sequence that directs the protein to the Golgi apparatus and which then remains uncleaved to function as a transmembrane anchor. By sequence similarity the beta4GalTs form four groups: beta4GalT1 and beta4GalT2 beta4GalT3 and beta4GalT4 beta4GalT5 and beta4GalT6 and beta4GalT7. The enzyme encoded by this gene appears to mainly play a role in glycolipid biosynthesis. Two alternatively spliced transcript variants have been found for this gene. Gene Aliases B4Gal-T4, beta4Gal-T4 RefSeq Accession No. NC_000003.11, NT_005612.16 UniGene ID Hs.13225 Ensembl Gene ID ENSG00000121578 Entrez Gene ID 8702 Assay Information Unique Assay ID qHsaCED0001445 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000483209, ENST00000467604, ENST00000471675, ENST00000359213, ENST00000393765, ENST00000475803, ENST00000479150 Amplicon Context Sequence TGAGGTAAGGAGACACAGAAGGGCAGTTGTCAAGTTCTACCTTCTTCGTGGATG CTTCATTAGTCAGAGTTTTTCCCTTCCCCAAAATGAGGGT Amplicon Length (bp) 64 Chromosome Location 3:118948694-118948787 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 103 Page 1/5 PrimePCR™Assay Validation Report R2 0.9998 cDNA Cq 21.23 cDNA Tm (Celsius) 78 gDNA Cq 23.9 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

MGAT5 Antibody (C-Term) Affinity Purified Rabbit Polyclonal Antibody (Pab) Catalog # Ap13815b
10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 MGAT5 Antibody (C-term) Affinity Purified Rabbit Polyclonal Antibody (Pab) Catalog # AP13815b Specification MGAT5 Antibody (C-term) - Product Information Application WB, FC,E Primary Accession Q09328 Other Accession NP_002401.1 Reactivity Human Host Rabbit Clonality Polyclonal Isotype Rabbit Ig Antigen Region 652-680 MGAT5 Antibody (C-term) - Additional Information Gene ID 4249 Other Names Alpha-1, 6-mannosylglycoprotein All lanes : Anti-MGAT5 Antibody (C-term) at 6-beta-N-acetylglucosaminyltransferase A, 1:1000 dilution Lane 1: HepG2 whole cell Alpha-mannoside beta-1, lysate Lane 2: Jurkat whole cell lysate 6-N-acetylglucosaminyltransferase, Lysates/proteins at 20 µg per lane. GlcNAc-T V, GNT-V, Mannoside Secondary Goat Anti-Rabbit IgG, (H+L), acetylglucosaminyltransferase 5, Peroxidase conjugated at 1/10000 dilution. N-acetylglucosaminyl-transferase V, MGAT5, GGNT5 Predicted band size : 85 kDa Blocking/Dilution buffer: 5% NFDM/TBST. Target/Specificity This MGAT5 antibody is generated from rabbits immunized with a KLH conjugated synthetic peptide between 652-680 amino acids from the C-terminal region of human MGAT5. Dilution WB~~1:1000 FC~~1:10~50 Format Purified polyclonal antibody supplied in PBS with 0.09% (W/V) sodium azide. This antibody is purified through a protein A column, followed by peptide affinity purification. MGAT5 Antibody (C-term) (Cat. #AP13815b) Storage flow cytometric analysis of NCI-H460 cells Maintain refrigerated at 2-8°C for up to 2 (right histogram) compared to a negative weeks. For long term storage store at -20°C control cell (left histogram).FITC-conjugated Page 1/3 10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 in small aliquots to prevent freeze-thaw donkey-anti-rabbit secondary antibodies were cycles. -

Mouse Mgat5 Knockout Project (CRISPR/Cas9)
https://www.alphaknockout.com Mouse Mgat5 Knockout Project (CRISPR/Cas9) Objective: To create a Mgat5 knockout Mouse model (C57BL/6J) by CRISPR/Cas-mediated genome engineering. Strategy summary: The Mgat5 gene (NCBI Reference Sequence: NM_145128 ; Ensembl: ENSMUSG00000036155 ) is located on Mouse chromosome 1. 18 exons are identified, with the ATG start codon in exon 3 and the TAG stop codon in exon 18 (Transcript: ENSMUST00000038361). Exon 3 will be selected as target site. Cas9 and gRNA will be co-injected into fertilized eggs for KO Mouse production. The pups will be genotyped by PCR followed by sequencing analysis. Note: Mice homozygous for deficiencies in this gene have immune system abnormalities and reduced cancer growth and metastasis. Exon 3 starts from the coding region. Exon 3 covers 10.86% of the coding region. The size of effective KO region: ~383 bp. The KO region does not have any other known gene. Page 1 of 9 https://www.alphaknockout.com Overview of the Targeting Strategy Wildtype allele gRNA region 5' gRNA region 3' 1 3 18 Legends Exon of mouse Mgat5 Knockout region Page 2 of 9 https://www.alphaknockout.com Overview of the Dot Plot (up) Window size: 15 bp Forward Reverse Complement Sequence 12 Note: The 2000 bp section upstream of Exon 3 is aligned with itself to determine if there are tandem repeats. Tandem repeats are found in the dot plot matrix. The gRNA site is selected outside of these tandem repeats. Overview of the Dot Plot (down) Window size: 15 bp Forward Reverse Complement Sequence 12 Note: The 2000 bp section downstream of Exon 3 is aligned with itself to determine if there are tandem repeats. -
![B4GALT4 Mouse Monoclonal Antibody (Biotin Conjugated) [Clone ID: OTI8B6] Product Data](https://docslib.b-cdn.net/cover/5706/b4galt4-mouse-monoclonal-antibody-biotin-conjugated-clone-id-oti8b6-product-data-1375706.webp)
B4GALT4 Mouse Monoclonal Antibody (Biotin Conjugated) [Clone ID: OTI8B6] Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for TA807809AM B4GALT4 Mouse Monoclonal Antibody (Biotin conjugated) [Clone ID: OTI8B6] Product data: Product Type: Primary Antibodies Clone Name: OTI8B6 Applications: IHC, WB Recommended Dilution: WB 1:500, IHC 1:150 Reactivity: Human Host: Mouse Isotype: IgG1 Clonality: Monoclonal Immunogen: Human recombinant protein fragment corresponding to amino acids 40-322 of human B4GALT4(NP_003769) produced in E.coli. Formulation: PBS (PH 7.3) containing 1% BSA, 50% glycerol and 0.02% sodium azide. Concentration: 0.5 mg/ml Purification: Purified from mouse ascites fluids or tissue culture supernatant by affinity chromatography (protein A/G) Conjugation: Biotin Storage: Store at -20°C as received. Stability: Stable for 12 months from date of receipt. Predicted Protein Size: 39.9 kDa Gene Name: beta-1,4-galactosyltransferase 4 Database Link: NP_003769 Entrez Gene 8702 Human O60513 This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 3 B4GALT4 Mouse Monoclonal Antibody (Biotin conjugated) [Clone ID: OTI8B6] – TA807809AM Background: This gene is one of seven beta-1,4-galactosyltransferase (beta4GalT) genes. They encode type II membrane-bound glycoproteins that appear to have exclusive specificity for the donor substrate UDP-galactose; all transfer galactose in a beta1,4 linkage to similar acceptor sugars: GlcNAc, Glc, and Xyl. Each beta4GalT has a distinct function in the biosynthesis of different glycoconjugates and saccharide structures. -

Same Role but Different Actors: Genetic Regulation of Post-Translational Modification of Two Distinct Proteins
bioRxiv preprint doi: https://doi.org/10.1101/2021.05.04.442584; this version posted May 4, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Same role but different actors: genetic regulation of post-translational modification of two distinct proteins Arianna Landini*1, Irena Trbojević-Akmačić*2, Pau Navarro3, Yakov A. Tsepilov4,5, Sodbo Z. Sharapov4, Frano Vučković2, Ozren Polašek6,7 Caroline Hayward3, Tea Petrovic2, Marija Vilaj2, Yurii S. Aulchenko4, Gordan Lauc*2,8, James F. Wilson*1,3, Lucija Klarić*3 1 Centre for Global Health Research, Usher Institute, University of Edinburgh, Edinburgh, United Kingdom 2 Genos Glycoscience Research Laboratory, Zagreb, Croatia 3 MRC Human Genetics Unit, Institute for Genetics and Cancer, University of Edinburgh, Edinburgh, United Kingdom 4 Laboratory of Glycogenomics, Institute of Cytology and Genetics, Novosibirsk, Russia 5 Laboratory of Theoretical and Applied Functional Genomics, Novosibirsk State University, Novosibirsk, Russia 6 Department of Public Health, School of Medicine, University of Split, Split, Croatia 7 Gen-info Ltd., Zagreb, Croatia 8 Faculty of Pharmacy and Biochemistry, University of Zagreb, Zagreb, Croatia * Authors contributed equally. Correspondence to: J.F.W ([email protected]) or L.K. ([email protected]) Post-translational modifications (PTMs) diversify protein functions and dynamically coordinate their signalling networks, influencing most aspects of cell physiology. Nevertheless, their genetic regulation or influence on complex traits is not fully understood. -

Duke University Dissertation Template
Gene-Environment Interactions in Cardiovascular Disease by Cavin Keith Ward-Caviness Graduate Program in Computational Biology and Bioinformatics Duke University Date:_______________________ Approved: ___________________________ Elizabeth R. Hauser, Supervisor ___________________________ William E. Kraus ___________________________ Sayan Mukherjee ___________________________ H. Frederik Nijhout Dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate Program in Computational Biology and Bioinformatics in the Graduate School of Duke University 2014 i v ABSTRACT Gene-Environment Interactions in Cardiovascular Disease by Cavin Keith Ward-Caviness Graduate Program in Computational Biology and Bioinformatics Duke University Date:_______________________ Approved: ___________________________ Elizabeth R. Hauser, Supervisor ___________________________ William E. Kraus ___________________________ Sayan Mukherjee ___________________________ H. Frederik Nijhout An abstract of a dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate Program in Computational Biology and Bioinformatics in the Graduate School of Duke University 2014 Copyright by Cavin Keith Ward-Caviness 2014 Abstract In this manuscript I seek to demonstrate the importance of gene-environment interactions in cardiovascular disease. This manuscript contains five studies each of which contributes to our understanding of the joint impact of genetic variation -

Genetic and Functional Approaches to Understanding Autoimmune and Inflammatory Pathologies
University of Vermont ScholarWorks @ UVM Graduate College Dissertations and Theses Dissertations and Theses 2020 Genetic And Functional Approaches To Understanding Autoimmune And Inflammatory Pathologies Abbas Raza University of Vermont Follow this and additional works at: https://scholarworks.uvm.edu/graddis Part of the Genetics and Genomics Commons, Immunology and Infectious Disease Commons, and the Pathology Commons Recommended Citation Raza, Abbas, "Genetic And Functional Approaches To Understanding Autoimmune And Inflammatory Pathologies" (2020). Graduate College Dissertations and Theses. 1175. https://scholarworks.uvm.edu/graddis/1175 This Dissertation is brought to you for free and open access by the Dissertations and Theses at ScholarWorks @ UVM. It has been accepted for inclusion in Graduate College Dissertations and Theses by an authorized administrator of ScholarWorks @ UVM. For more information, please contact [email protected]. GENETIC AND FUNCTIONAL APPROACHES TO UNDERSTANDING AUTOIMMUNE AND INFLAMMATORY PATHOLOGIES A Dissertation Presented by Abbas Raza to The Faculty of the Graduate College of The University of Vermont In Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy Specializing in Cellular, Molecular, and Biomedical Sciences January, 2020 Defense Date: August 30, 2019 Dissertation Examination Committee: Cory Teuscher, Ph.D., Advisor Jonathan Boyson, Ph.D., Chairperson Matthew Poynter, Ph.D. Ralph Budd, M.D. Dawei Li, Ph.D. Dimitry Krementsov, Ph.D. Cynthia J. Forehand, Ph.D., Dean of the Graduate College ABSTRACT Our understanding of genetic predisposition to inflammatory and autoimmune diseases has been enhanced by large scale quantitative trait loci (QTL) linkage mapping and genome-wide association studies (GWAS). However, the resolution and interpretation of QTL linkage mapping or GWAS findings are limited. -

Identification of Novel Cell Glycolysis Related Gene Signature Predicting
Wang et al. Cancer Cell Int (2019) 19:296 https://doi.org/10.1186/s12935-019-1001-0 Cancer Cell International PRIMARY RESEARCH Open Access Identifcation of novel cell glycolysis related gene signature predicting survival in patients with endometrial cancer Zi‑Hao Wang, Yun‑Zheng Zhang, Yu‑Shan Wang and Xiao‑Xin Ma* Abstract Background: Endometrial cancer (EC) is one of the three major gynecological malignancies. Numerous biomarkers that may be associated with survival and prognosis have been identifed through database mining in previous stud‑ ies. However, the predictive ability of single‑gene biomarkers is not sufciently specifc. Genetic signatures may be an improved option for prediction. This study aimed to explore data from The Cancer Genome Atlas (TCGA) to identify a new genetic signature for predicting the prognosis of EC. Methods: mRNA expression profling was performed in a group of patients with EC (n 548) from TCGA. Gene set enrichment analysis was performed to identify gene sets that were signifcantly diferent= between EC tissues and normal tissues. Cox proportional hazards regression models were used to identify genes signifcantly associated with overall survival. Quantitative real‑time‑PCR was used to verify the reliability of the expression of selected mRNAs. Subsequent multivariate Cox regression analysis was used to establish a prognostic risk parameter formula. Kaplan– Meier survival estimates and the log‐rank test were used to validate the signifcance of risk parameters for prognosis prediction. Result: Nine genes associated with glycolysis (CLDN9, B4GALT1, GMPPB, B4GALT4, AK4, CHST6, PC, GPC1, and SRD5A3) were found to be signifcantly related to overall survival. The results of mRNA expression analysis by PCR were consist‑ ent with those of bioinformatics analysis. -

A Network Based Approach to Identify the Genetic Influence Caused By
bioRxiv preprint doi: https://doi.org/10.1101/482760; this version posted November 30, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. A Network based Approach to Identify the Genetic Influence Caused by Associated Factors and Disorders for the Parkinsons Disease Progression 1st Najmus Sakib 1st Utpala Nanda Chowdhury Dept. of Applied Physics and Electronic Engineering Dept. of Computer Science and Engineering University of Rajshahi University of Rajshahi Rakshahi, Bangladesh Rakshahi, Bangladesh najmus:sakib1995@outlook:com unchowdhury@ru:ac:bd 2nd M. Babul Islam 3rd Julian M.W. Quinn 4th Mohammad Ali Moni Dept. of Applied Physics and Electronic Engg. Bone biology divisions School of Medical Science University of Rajshahi Garvan Institute of Medical Research The University of Sydney Rakshahi, Bangladesh NSW 2010 NSW, Australia babul:apee@ru:ac:bd j:quinn@garvan:org:au mohammad:moni@sydney:edu:au Abstract—Actual causes of Parkinsons disease (PD) are still the central nervous system [1]. It is one of the most common unknown. In any case, a better comprehension of genetic and neurodegenerative problems after Alzheimers illness all over ecological influences to the PD and their interaction will assist the world [2]. The PD is characterized by progressive damage physicians and patients to evaluate individual hazard for the PD, and definitely, there will be a possibility to find a way to of dopaminergic neurons in the substantia nigra pars compacta reduce the progression of the PD.