Characteristics and Prognostic Value of Potential Dependency Genes In
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Seq2pathway Vignette
seq2pathway Vignette Bin Wang, Xinan Holly Yang, Arjun Kinstlick May 19, 2021 Contents 1 Abstract 1 2 Package Installation 2 3 runseq2pathway 2 4 Two main functions 3 4.1 seq2gene . .3 4.1.1 seq2gene flowchart . .3 4.1.2 runseq2gene inputs/parameters . .5 4.1.3 runseq2gene outputs . .8 4.2 gene2pathway . 10 4.2.1 gene2pathway flowchart . 11 4.2.2 gene2pathway test inputs/parameters . 11 4.2.3 gene2pathway test outputs . 12 5 Examples 13 5.1 ChIP-seq data analysis . 13 5.1.1 Map ChIP-seq enriched peaks to genes using runseq2gene .................... 13 5.1.2 Discover enriched GO terms using gene2pathway_test with gene scores . 15 5.1.3 Discover enriched GO terms using Fisher's Exact test without gene scores . 17 5.1.4 Add description for genes . 20 5.2 RNA-seq data analysis . 20 6 R environment session 23 1 Abstract Seq2pathway is a novel computational tool to analyze functional gene-sets (including signaling pathways) using variable next-generation sequencing data[1]. Integral to this tool are the \seq2gene" and \gene2pathway" components in series that infer a quantitative pathway-level profile for each sample. The seq2gene function assigns phenotype-associated significance of genomic regions to gene-level scores, where the significance could be p-values of SNPs or point mutations, protein-binding affinity, or transcriptional expression level. The seq2gene function has the feasibility to assign non-exon regions to a range of neighboring genes besides the nearest one, thus facilitating the study of functional non-coding elements[2]. Then the gene2pathway summarizes gene-level measurements to pathway-level scores, comparing the quantity of significance for gene members within a pathway with those outside a pathway. -

Supplementary Table 2 Gene Sets Used in GSEA
Supplementary Table 2 Gene sets used in GSEA Up in RNAi and Sign Confirmed in Inducible Gene Probe Set ID Accession Symbol Gene Title 200660_at NM_005620 S100A11 S100 calcium binding protein A11 (calgizzarin) 200785_s_at NM_002332 LRP1 low density lipoprotein-related protein 1 (alpha-2-macroglobulin receptor) 201325_s_at NM_001423 EMP1 epithelial membrane protein 1 201373_at NM_000445 PLEC1 plectin 1, intermediate filament binding protein 500kDa 201466_s_at NM_002228 JUN v-jun sarcoma virus 17 oncogene homolog (avian) 201952_at AA156721 ALCAM activated leukocyte cell adhesion molecule 202042_at NM_002109 HARS histidyl-tRNA synthetase 202074_s_at NM_021980 OPTN optineurin 202087_s_at NM_001912 CTSL cathepsin L 202588_at NM_000476 AK1 adenylate kinase 1 202609_at NM_004447 EPS8 epidermal growth factor receptor pathway substrate 8 202733_at NM_004199 P4HA2 procollagen-proline, 2-oxoglutarate 4-dioxygenase (proline 4-hydroxylase), alpha polypeptide II 202756_s_at NM_002081 GPC1 glypican 1 202786_at NM_013233 STK39 serine threonine kinase 39 (STE20/SPS1 homolog, yeast) 202859_x_at NM_000584 IL8 interleukin 8 203083_at NM_003247 THBS2 thrombospondin 2 203186_s_at NM_002961 S100A4 S100 calcium binding protein A4 (calcium protein, calvasculin, metastasin, murine placental homolog) 203232_s_at NM_000332 ATXN1 ataxin 1 203233_at NM_000418 IL4R interleukin 4 receptor 203771_s_at AA740186 BLVRA biliverdin reductase A 203821_at NM_001945 HBEGF heparin-binding EGF-like growth factor 203939_at NM_002526 NT5E 5'-nucleotidase, ecto (CD73) 203955_at NM_014811 -
Download Validation Data
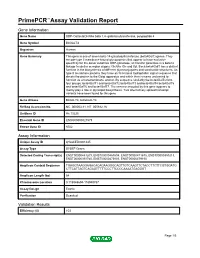
PrimePCR™Assay Validation Report Gene Information Gene Name UDP-Gal:betaGlcNAc beta 1,4- galactosyltransferase, polypeptide 4 Gene Symbol B4GALT4 Organism Human Gene Summary This gene is one of seven beta-14-galactosyltransferase (beta4GalT) genes. They encode type II membrane-bound glycoproteins that appear to have exclusive specificity for the donor substrate UDP-galactose; all transfer galactose in a beta14 linkage to similar acceptor sugars: GlcNAc Glc and Xyl. Each beta4GalT has a distinct function in the biosynthesis of different glycoconjugates and saccharide structures. As type II membrane proteins they have an N-terminal hydrophobic signal sequence that directs the protein to the Golgi apparatus and which then remains uncleaved to function as a transmembrane anchor. By sequence similarity the beta4GalTs form four groups: beta4GalT1 and beta4GalT2 beta4GalT3 and beta4GalT4 beta4GalT5 and beta4GalT6 and beta4GalT7. The enzyme encoded by this gene appears to mainly play a role in glycolipid biosynthesis. Two alternatively spliced transcript variants have been found for this gene. Gene Aliases B4Gal-T4, beta4Gal-T4 RefSeq Accession No. NC_000003.11, NT_005612.16 UniGene ID Hs.13225 Ensembl Gene ID ENSG00000121578 Entrez Gene ID 8702 Assay Information Unique Assay ID qHsaCED0001445 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000483209, ENST00000467604, ENST00000471675, ENST00000359213, ENST00000393765, ENST00000475803, ENST00000479150 Amplicon Context Sequence TGAGGTAAGGAGACACAGAAGGGCAGTTGTCAAGTTCTACCTTCTTCGTGGATG CTTCATTAGTCAGAGTTTTTCCCTTCCCCAAAATGAGGGT Amplicon Length (bp) 64 Chromosome Location 3:118948694-118948787 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 103 Page 1/5 PrimePCR™Assay Validation Report R2 0.9998 cDNA Cq 21.23 cDNA Tm (Celsius) 78 gDNA Cq 23.9 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -
![B4GALT4 Mouse Monoclonal Antibody (Biotin Conjugated) [Clone ID: OTI8B6] Product Data](https://docslib.b-cdn.net/cover/5706/b4galt4-mouse-monoclonal-antibody-biotin-conjugated-clone-id-oti8b6-product-data-1375706.webp)
B4GALT4 Mouse Monoclonal Antibody (Biotin Conjugated) [Clone ID: OTI8B6] Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for TA807809AM B4GALT4 Mouse Monoclonal Antibody (Biotin conjugated) [Clone ID: OTI8B6] Product data: Product Type: Primary Antibodies Clone Name: OTI8B6 Applications: IHC, WB Recommended Dilution: WB 1:500, IHC 1:150 Reactivity: Human Host: Mouse Isotype: IgG1 Clonality: Monoclonal Immunogen: Human recombinant protein fragment corresponding to amino acids 40-322 of human B4GALT4(NP_003769) produced in E.coli. Formulation: PBS (PH 7.3) containing 1% BSA, 50% glycerol and 0.02% sodium azide. Concentration: 0.5 mg/ml Purification: Purified from mouse ascites fluids or tissue culture supernatant by affinity chromatography (protein A/G) Conjugation: Biotin Storage: Store at -20°C as received. Stability: Stable for 12 months from date of receipt. Predicted Protein Size: 39.9 kDa Gene Name: beta-1,4-galactosyltransferase 4 Database Link: NP_003769 Entrez Gene 8702 Human O60513 This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 3 B4GALT4 Mouse Monoclonal Antibody (Biotin conjugated) [Clone ID: OTI8B6] – TA807809AM Background: This gene is one of seven beta-1,4-galactosyltransferase (beta4GalT) genes. They encode type II membrane-bound glycoproteins that appear to have exclusive specificity for the donor substrate UDP-galactose; all transfer galactose in a beta1,4 linkage to similar acceptor sugars: GlcNAc, Glc, and Xyl. Each beta4GalT has a distinct function in the biosynthesis of different glycoconjugates and saccharide structures. -

Duke University Dissertation Template
Gene-Environment Interactions in Cardiovascular Disease by Cavin Keith Ward-Caviness Graduate Program in Computational Biology and Bioinformatics Duke University Date:_______________________ Approved: ___________________________ Elizabeth R. Hauser, Supervisor ___________________________ William E. Kraus ___________________________ Sayan Mukherjee ___________________________ H. Frederik Nijhout Dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate Program in Computational Biology and Bioinformatics in the Graduate School of Duke University 2014 i v ABSTRACT Gene-Environment Interactions in Cardiovascular Disease by Cavin Keith Ward-Caviness Graduate Program in Computational Biology and Bioinformatics Duke University Date:_______________________ Approved: ___________________________ Elizabeth R. Hauser, Supervisor ___________________________ William E. Kraus ___________________________ Sayan Mukherjee ___________________________ H. Frederik Nijhout An abstract of a dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate Program in Computational Biology and Bioinformatics in the Graduate School of Duke University 2014 Copyright by Cavin Keith Ward-Caviness 2014 Abstract In this manuscript I seek to demonstrate the importance of gene-environment interactions in cardiovascular disease. This manuscript contains five studies each of which contributes to our understanding of the joint impact of genetic variation -

Identification of Novel Cell Glycolysis Related Gene Signature Predicting
Wang et al. Cancer Cell Int (2019) 19:296 https://doi.org/10.1186/s12935-019-1001-0 Cancer Cell International PRIMARY RESEARCH Open Access Identifcation of novel cell glycolysis related gene signature predicting survival in patients with endometrial cancer Zi‑Hao Wang, Yun‑Zheng Zhang, Yu‑Shan Wang and Xiao‑Xin Ma* Abstract Background: Endometrial cancer (EC) is one of the three major gynecological malignancies. Numerous biomarkers that may be associated with survival and prognosis have been identifed through database mining in previous stud‑ ies. However, the predictive ability of single‑gene biomarkers is not sufciently specifc. Genetic signatures may be an improved option for prediction. This study aimed to explore data from The Cancer Genome Atlas (TCGA) to identify a new genetic signature for predicting the prognosis of EC. Methods: mRNA expression profling was performed in a group of patients with EC (n 548) from TCGA. Gene set enrichment analysis was performed to identify gene sets that were signifcantly diferent= between EC tissues and normal tissues. Cox proportional hazards regression models were used to identify genes signifcantly associated with overall survival. Quantitative real‑time‑PCR was used to verify the reliability of the expression of selected mRNAs. Subsequent multivariate Cox regression analysis was used to establish a prognostic risk parameter formula. Kaplan– Meier survival estimates and the log‐rank test were used to validate the signifcance of risk parameters for prognosis prediction. Result: Nine genes associated with glycolysis (CLDN9, B4GALT1, GMPPB, B4GALT4, AK4, CHST6, PC, GPC1, and SRD5A3) were found to be signifcantly related to overall survival. The results of mRNA expression analysis by PCR were consist‑ ent with those of bioinformatics analysis. -

Identifying Rare-Variant Associations in Parent-Child Trios Using a Gaussian
Lu and Cantor BMC Proceedings 2014, 8(Suppl 1):S98 http://www.biomedcentral.com/1753-6561/8/S1/S98 PROCEEDINGS Open Access Identifying rare-variant associations in parent-child trios using a Gaussian support vector machine Ake T Lu1, Rita M Cantor1,2* From Genetic Analysis Workshop 18 Stevenson, WA, USA. 13-17 October 2012 Abstract As the availability of cost-effective high-throughput sequencing technology increases, genetic research is beginning to focus on identifying the contributions of rare variants (RVs) to complex traits. Using RVs to detect associated genes requires statistical approaches that mitigate the lack of power with the analysis of single RVs. Here we report the development and application of an approach that aggregates and evaluates the transmissions of RVs in parent- child trios. An initial score that incorporates the distortion in transmission of the observed RVs from the parents to their offspring is calculated for each variant. The scores are analyzed using a support vector machine that handles these data by mapping the transmission distortion of the multiple RVs into a one-dimensional score in a nonlinear fashion when parent-child trios with affected and nonaffected children are contrasted. We refer to this approach as Trio-SVM. A total of 275 trios were available in the Genetic Analysis Workshop 18 data for analysis. Because of their nonindependence and the extended linkage disequilibrium (LD) within pedigrees, Trio-SVM was vulnerable to type I errors in detecting association. Using the GAW18 data with simulated trait values, Trio-SVM has an appropriate type I error, but it lacks power with a sample of 267 trios. -

B4GALT4 Human Sirna Oligo Duplex (Locus ID 8702) Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for SR305726 B4GALT4 Human siRNA Oligo Duplex (Locus ID 8702) Product data: Product Type: siRNA Oligo Duplexes Purity: HPLC purified Quality Control: Tested by ESI-MS Sequences: Available with shipment Stability: One year from date of shipment when stored at -20°C. # of transfections: Approximately 330 transfections/2nmol in 24-well plate under optimized conditions (final conc. 10 nM). Note: Single siRNA duplex (10nmol) can be ordered. RefSeq: NM_003778, NM_212543 Synonyms: B4Gal-T4; beta4Gal-T4 Components: B4GALT4 (Human) - 3 unique 27mer siRNA duplexes - 2 nmol each (Locus ID 8702) Included - SR30004, Trilencer-27 Universal Scrambled Negative Control siRNA Duplex - 2 nmol Included - SR30005, RNAse free siRNA Duplex Resuspension Buffer - 2 ml Summary: This gene is one of seven beta-1,4-galactosyltransferase (beta4GalT) genes. They encode type II membrane-bound glycoproteins that appear to have exclusive specificity for the donor substrate UDP-galactose; all transfer galactose in a beta1,4 linkage to similar acceptor sugars: GlcNAc, Glc, and Xyl. Each beta4GalT has a distinct function in the biosynthesis of different glycoconjugates and saccharide structures. As type II membrane proteins, they have an N- terminal hydrophobic signal sequence that directs the protein to the Golgi apparatus and which then remains uncleaved to function as a transmembrane anchor. By sequence similarity, the beta4GalTs form four groups: beta4GalT1 and beta4GalT2, beta4GalT3 and beta4GalT4, beta4GalT5 and beta4GalT6, and beta4GalT7. The enzyme encoded by this gene appears to mainly play a role in glycolipid biosynthesis. -

Multiplexed Surrogate Analysis of Glycotransferase Activity in Whole Biospecimens † † ‡ Chad R
Article pubs.acs.org/ac Multiplexed Surrogate Analysis of Glycotransferase Activity in Whole Biospecimens † † ‡ Chad R. Borges, ,* Douglas S. Rehder, and Paolo Boffetta † Molecular Biomarkers Unit, The Biodesign Institute at Arizona State University, Tempe, Arizona 85287, United States ‡ Institute for Translational Epidemiology and Tisch Cancer Institute, Mount Sinai School of Medicine, New York, New York 10029, United States *S Supporting Information ABSTRACT: Dysregulated glycotransferase enzymes in can- cer cells produce aberrant glycanssome of which can help facilitate metastases. Within a cell, individual glycotransferases promiscuously help to construct dozens of unique glycan structures, making it difficult to comprehensively track their activity in biospecimensespecially where they are absent or inactive. Here, we describe an approach to deconstruct glycans in whole biospecimens then analytically pool together resulting monosaccharide-and-linkage-specific degradation products (“glycan nodes”) that directly represent the activities of specific glycotransferases. To implement this concept, a reproducible, relative quantitation-based glycan methylation analysis methodology was developed that simultaneously captures information from N-, O-, and lipid linked glycans and is compatible with whole biofluids and homogenized tissues; in total, over 30 different glycan nodes are detectable per gas chromatography−mass spectrometry (GC-MS) run. Numerous nonliver organ cancers are known to induce the production of abnormally glycosylated serum proteins. Thus, following analytical validation, in blood plasma, the technique was applied to a group of 59 lung cancer patient plasma samples and age/gender/ smoking-status-matched non-neoplastic controls from the Lung Cancer in Central and Eastern Europe (CEE) study to gauge the clinical utility of the approach toward the detection of lung cancer. -

Acetyl-Coa Flux from the Cytosol to the ER Regulates Engagement And
www.nature.com/scientificreports OPEN Acetyl‑CoA fux from the cytosol to the ER regulates engagement and quality of the secretory pathway Inca A. Dieterich1,2,3,11, Yusi Cui4,11, Megan M. Braun1,2,3, Alexis J. Lawton5,6, Nicklaus H. Robinson1,2, Jennifer L. Peotter5, Qing Yu4,10, Jason C. Casler7, Benjamin S. Glick7, Anjon Audhya5, John M. Denu5,6, Lingjun Li4* & Luigi Puglielli1,2,8,9* Nε‑lysine acetylation in the ER is an essential component of the quality control machinery. ER acetylation is ensured by a membrane transporter, AT‑1/SLC33A1, which translocates cytosolic acetyl‑ CoA into the ER lumen, and two acetyltransferases, ATase1 and ATase2, which acetylate nascent polypeptides within the ER lumen. Dysfunctional AT‑1, as caused by gene mutation or duplication events, results in severe disease phenotypes. Here, we used two models of AT‑1 dysregulation to investigate dynamics of the secretory pathway: AT‑1 sTg, a model of systemic AT‑1 overexpression, and AT‑1S113R/+, a model of AT‑1 haploinsufciency. The animals displayed reorganization of the ER, ERGIC, and Golgi apparatus. In particular, AT‑1 sTg animals displayed a marked delay in Golgi‑to‑ plasma membrane protein trafcking, signifcant alterations in Golgi‑based N‑glycan modifcation, and a marked expansion of the lysosomal network. Collectively our results indicate that AT‑1 is essential to maintain proper organization and engagement of the secretory pathway. Nε-lysine acetylation in the Endoplasmic Reticulum (ER) has emerged as an essential component of the qual- ity control (QC) machinery that maintains protein homeostasis (proteostasis) within the ER1–6. -

Bioinformatics-Based Prediction of FUT8 As a Therapeutic Target in Estrogen Receptor-Positive Breast Cancer Fateme Shaabanpour Aghamaleki1, Shirin Farivar1,*
Multidisciplinary Cancer Investigation Original Article January 2019, Volume 3, Issue 1 Bioinformatics-Based Prediction of FUT8 as a Therapeutic Target in Estrogen Receptor-Positive Breast Cancer Fateme Shaabanpour Aghamaleki1, Shirin Farivar1,* 1 Department of Cellullar-Molecular Biology , Faculty of biological Sciences and technologies, Shahid Beheshti University G.C., Tehran, Iran *Corresponding author: Shirin Farivar,Department of Cellullar-Molecular Biology, Faculty of biological Sciences and technologies, Shahid Beheshti University G.C., Tehran, Iran. Tel: +982129902720; Fax: +982122431664; E-mail: [email protected] DOI: 10.30699/acadpub.mci.3.1.25 Abstract Submitted: 20 October 2018 Introduction: Estrogen receptor-positive (ER-positive) breast cancer is a subgroup of Revised: 4 November 2018 breast tumors that is more likely to respond to hormone therapy. ER-positive and ER- Accepted: 28 November 2018 negative breast cancers tend to show different patterns of metastasis because of different e-Published: 1 January 2019 signaling cascade and genes that are activated by estrogen response. Genetic factors can contribute to high rates of metastasis in ER-positive breast cancer. Fucosyltransferase 8 Keywords: (FUT8) is a member of fucosyltransferases family and plays an important role in α-1,6 Fucosyltransferases linkage to the first GlcNAc residue of N-glycans chain. In this study, for the first time, Breast Neoplasms Microarray Analysis we predicted FUT8 by bioinformatics tools as a novel therapeutic target for ER-positive Signal Transduction breast cancer. Methods: Microarray gene expression data of 9 patients with ER+ve and 10 individuals with ER-ve breast cancer was extracted from Geodatasets. Gene expression of two ER+ and ER- patients was compared with logfc and then sorted by their p-values.