Mutations of NTRK3, EPHA3 and GUCY2F in Human Cancers
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

GUCY2F Protein Recombinant Human Protein Expressed in Sf9 Cells
Catalog # Aliquot Size G12-31G-20 20 µg G12-31G-50 50 µg GUCY2F Protein Recombinant human protein expressed in sf9 cells Catalog # G12-31G Lot # U1858-3 Product Description Purity Recombinant human GUCY2F (367-end) was expressed by baculovirus in Sf9 insect cells using an N-terminal GST tag. The gene accession number is BC156674. The purity of GUCY2F protein was determined to be >70% by Gene Aliases densitometry, GUCY2F approx. MW 110kDa. CYGF, GC-F, GUC2DL, GUC2F, RETGC-2, ROS-GC2 Formulation Recombinant protein stored in 50mM Tris-HCl, pH 7.5, 150mM NaCl, 10mM glutathione, 0.1mM EDTA, 0.25mM DTT, 0.1mM PMSF, 25% glycerol. Storage and Stability Store product at –70oC. For optimal storage, aliquot target into smaller quantities after centrifugation and store at recommended temperature. For most favorable performance, avoid repeated handling and multiple freeze/thaw cycles. Scientific Background Guanylate cyclase 2F (GUCY2F) belongs to the adenylyl cyclase class-4/guanylyl cyclase family which also includes GUCY2D. Both GUCY2F and GUCY2D are responsible for the replenishment of cGMP in photoreceptors after exposure to light and are required for the normal kinetics of photoreceptor sensitivity and recovery, although disease mutations are restricted to GUCY2F Protein Full-length recombinant human protein expressed in sf9 cells GUCY2D. Catalog # G12-31G References Lot # U1858-3 1. Goraczniak R, et al: Structural and functional characteri- Purity >70% Concentration 0.05 µg/µl zation of a second subfamily member of the calcium- Stability 1yr at –70oC from date of shipment modulated bovine rod outer segment membrane Storage & Shipping Store product at –70oC. -

Supplementary Table 2
Supplementary Table 2. Differentially Expressed Genes following Sham treatment relative to Untreated Controls Fold Change Accession Name Symbol 3 h 12 h NM_013121 CD28 antigen Cd28 12.82 BG665360 FMS-like tyrosine kinase 1 Flt1 9.63 NM_012701 Adrenergic receptor, beta 1 Adrb1 8.24 0.46 U20796 Nuclear receptor subfamily 1, group D, member 2 Nr1d2 7.22 NM_017116 Calpain 2 Capn2 6.41 BE097282 Guanine nucleotide binding protein, alpha 12 Gna12 6.21 NM_053328 Basic helix-loop-helix domain containing, class B2 Bhlhb2 5.79 NM_053831 Guanylate cyclase 2f Gucy2f 5.71 AW251703 Tumor necrosis factor receptor superfamily, member 12a Tnfrsf12a 5.57 NM_021691 Twist homolog 2 (Drosophila) Twist2 5.42 NM_133550 Fc receptor, IgE, low affinity II, alpha polypeptide Fcer2a 4.93 NM_031120 Signal sequence receptor, gamma Ssr3 4.84 NM_053544 Secreted frizzled-related protein 4 Sfrp4 4.73 NM_053910 Pleckstrin homology, Sec7 and coiled/coil domains 1 Pscd1 4.69 BE113233 Suppressor of cytokine signaling 2 Socs2 4.68 NM_053949 Potassium voltage-gated channel, subfamily H (eag- Kcnh2 4.60 related), member 2 NM_017305 Glutamate cysteine ligase, modifier subunit Gclm 4.59 NM_017309 Protein phospatase 3, regulatory subunit B, alpha Ppp3r1 4.54 isoform,type 1 NM_012765 5-hydroxytryptamine (serotonin) receptor 2C Htr2c 4.46 NM_017218 V-erb-b2 erythroblastic leukemia viral oncogene homolog Erbb3 4.42 3 (avian) AW918369 Zinc finger protein 191 Zfp191 4.38 NM_031034 Guanine nucleotide binding protein, alpha 12 Gna12 4.38 NM_017020 Interleukin 6 receptor Il6r 4.37 AJ002942 -

Research Article Complex and Multidimensional Lipid Raft Alterations in a Murine Model of Alzheimer’S Disease
SAGE-Hindawi Access to Research International Journal of Alzheimer’s Disease Volume 2010, Article ID 604792, 56 pages doi:10.4061/2010/604792 Research Article Complex and Multidimensional Lipid Raft Alterations in a Murine Model of Alzheimer’s Disease Wayne Chadwick, 1 Randall Brenneman,1, 2 Bronwen Martin,3 and Stuart Maudsley1 1 Receptor Pharmacology Unit, National Institute on Aging, National Institutes of Health, 251 Bayview Boulevard, Suite 100, Baltimore, MD 21224, USA 2 Miller School of Medicine, University of Miami, Miami, FL 33124, USA 3 Metabolism Unit, National Institute on Aging, National Institutes of Health, 251 Bayview Boulevard, Suite 100, Baltimore, MD 21224, USA Correspondence should be addressed to Stuart Maudsley, [email protected] Received 17 May 2010; Accepted 27 July 2010 Academic Editor: Gemma Casadesus Copyright © 2010 Wayne Chadwick et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Various animal models of Alzheimer’s disease (AD) have been created to assist our appreciation of AD pathophysiology, as well as aid development of novel therapeutic strategies. Despite the discovery of mutated proteins that predict the development of AD, there are likely to be many other proteins also involved in this disorder. Complex physiological processes are mediated by coherent interactions of clusters of functionally related proteins. Synaptic dysfunction is one of the hallmarks of AD. Synaptic proteins are organized into multiprotein complexes in high-density membrane structures, known as lipid rafts. These microdomains enable coherent clustering of synergistic signaling proteins. -
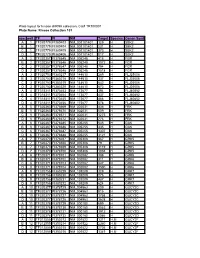
Kinase Collection 101 Row Well TF FI NM Target Location Spec
Plate layout for kinase shRNA collection, Cat# TR100001 Plate Name: Kinase Collection 101 row well TF FI NM Target LocationSpecies Genen Symbol A 1 TF320779 FI380403 NM_001101401 129 H SBK2 B 1 TF320779 FI380404 NM_001101401 231 H SBK2 C 1 TF320779 FI380405 NM_001101401 373 H SBK2 D 1 TF320779 FI380406 NM_001101401 817 H SBK2 A 2 TF320357 FI378645 NM_005248 415 H FGR B 2 TF320357 FI378646 NM_005248 1072 H FGR C 2 TF320357 FI378647 NM_005248 794 H FGR D 2 TF320357 FI378648 NM_005248 1518 H FGR A 3 TF320753 FI380217 NM_144610 289 H FLJ25006 B 3 TF320753 FI380218 NM_144610 137 H FLJ25006 C 3 TF320753 FI380219 NM_144610 642 H FLJ25006 D 3 TF320753 FI380220 NM_144610 673 H FLJ25006 A 4 TF318311 FI370453 NM_173677 394 H FLJ40852 B 4 TF318311 FI370454 NM_173677 437 H FLJ40852 C 4 TF318311 FI370455 NM_173677 466 H FLJ40852 D 4 TF318311 FI370456 NM_173677 578 H FLJ40852 A 5 TF320363 FI378669 NM_002031 436 H FRK B 5 TF320363 FI378670 NM_002031 509 H FRK C 5 TF320363 FI378671 NM_002031 1275 H FRK D 5 TF320363 FI378672 NM_002031 776 H FRK A 6 TF320367 FI378685 NM_005255 545 H GAK B 6 TF320367 FI378686 NM_005255 435 H GAK C 6 TF320367 FI378687 NM_005255 1307 H GAK D 6 TF320367 FI378688 NM_005255 3121 H GAK A 7 TF320370 FI378697 NM_005308 367 H GRK5 B 7 TF320370 FI378698 NM_005308 79 H GRK5 C 7 TF320370 FI378699 NM_005308 1119 H GRK5 D 7 TF320370 FI378700 NM_005308 1038 H GRK5 A 8 TF320371 FI378701 NM_002082 388 H GRK6 B 8 TF320371 FI378702 NM_002082 311 H GRK6 C 8 TF320371 FI378703 NM_002082 847 H GRK6 D 8 TF320371 FI378704 NM_002082 1590 H GRK6 A -

Expression Profiles of Three Novel Sensory Guanylate Cyclases and Guanylate Cyclase-Activating Proteins in the Zebrafish Retina
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Biochimica et Biophysica Acta 1793 (2009) 1110–1114 Contents lists available at ScienceDirect Biochimica et Biophysica Acta journal homepage: www.elsevier.com/locate/bbamcr Expression profiles of three novel sensory guanylate cyclases and guanylate cyclase-activating proteins in the zebrafish retina Nina Rätscho, Alexander Scholten, Karl-Wilhelm Koch ⁎ Biochemistry Group, Institute of Biology and Environmental Science, Faculty V, Carl von Ossietzky University Oldenburg, D-26111 Oldenburg, Germany article info abstract Article history: Three membrane bound sensory guanylate cyclases are expressed in photoreceptor cells of the developing Received 30 September 2008 and adult zebrafish retina. First appearance of mRNA transcripts was detected by in situ hybridization Received in revised form 19 November 2008 techniques for all guanylate cyclases between 3 and 4 days post fertilization (dpf), but only one isoform Accepted 27 December 2008 (guanylate cyclase 3) appeared to be specifically expressed in cones of the adult retina. Transcripts of three Available online 6 January 2009 cone specific guanylate cyclase-activating proteins (zGCAP3, zGCAP4 and zGCAP7) were also detected at 3– Keywords: 4 dpf. The expression onset of the guanylate cyclases and these neuronal calcium sensor proteins mainly Guanylate cyclase overlapped. High guanylate cyclase activities in larval eye preparations and the precisely controlled GCAP coexpression of guanylate cyclases and zGCAPs coincide with the onset of visual function at 3–4 dpf. Neuronal calcium sensor protein © 2009 Elsevier B.V. All rights reserved. Retina 1. Introduction of the full-length sequences and a characterization of their molecular properties has not been reported so far. -

Network-Based Analysis of Key Regulatory Genes Implicated in Type
www.nature.com/scientificreports OPEN Network‑based analysis of key regulatory genes implicated in Type 2 Diabetes Mellitus and Recurrent Miscarriages in Turner Syndrome Anam Farooqui1, Alaa Alhazmi2, Shaful Haque3, Naaila Tamkeen4, Mahboubeh Mehmankhah1, Safa Tazyeen1, Sher Ali5 & Romana Ishrat1* The information on the genotype–phenotype relationship in Turner Syndrome (TS) is inadequate because very few specifc candidate genes are linked to its clinical features. We used the microarray data of TS to identify the key regulatory genes implicated with TS through a network approach. The causative factors of two common co‑morbidities, Type 2 Diabetes Mellitus (T2DM) and Recurrent Miscarriages (RM), in the Turner population, are expected to be diferent from that of the general population. Through microarray analysis, we identifed nine signature genes of T2DM and three signature genes of RM in TS. The power‑law distribution analysis showed that the TS network carries scale‑free hierarchical fractal attributes. Through local‑community‑paradigm (LCP) estimation we fnd that a strong LCP is also maintained which means that networks are dynamic and heterogeneous. We identifed nine key regulators which serve as the backbone of the TS network. Furthermore, we recognized eight interologs functional in seven diferent organisms from lower to higher levels. Overall, these results ofer few key regulators and essential genes that we envisage have potential as therapeutic targets for the TS in the future and the animal models studied here may prove useful in the validation of such targets. Te medical systems and scientists throughout the world are under an unprecedented challenge to meet the medical needs of much of the world’s population that are sufering from chromosomal anomalies. -

Gucy2f Zebrafish Knockdown &Ndash; a Model for Gucy2d-Related
European Journal of Human Genetics (2012) 20, 884–889 & 2012 Macmillan Publishers Limited All rights reserved 1018-4813/12 www.nature.com/ejhg ARTICLE Gucy2f zebrafish knockdown – a model for Gucy2d-related leber congenital amaurosis Hadas Stiebel-Kalish*,1,2,3, Ehud Reich2,3,8, Nir Rainy4,8, Gad Vatine4, Yael Nisgav5, Anna Tovar6, Yoav Gothilf4 and Michael Bach7 Mutations in retinal-specific guanylate cyclase (Gucy2d) are associated with Leber congenital amaurosis-1 (LCA1). Zebrafish offer unique advantages relative to rodents, including their excellent color vision, precocious retinal development, robust visual testing strategies, low cost, relatively easy transgenesis and shortened experimental times. In this study we will demonstrate the feasibility of using gene-targeting in the zebrafish as a model for the photoreceptor-specific GUCY2D-related LCA1, by reporting the visual phenotype and retinal histology resulting from Gucy2f knockdown. Gucy2f zebrafish LCA-orthologous cDNA was identified and isolated by PCR amplification. Its expression pattern was determined by whole-mount in-situ hybridization and its function was studied by gene knockdown using two different morpholino-modified oligos (MO), one that blocks translation of Gucy2f and one that blocks splicing of Gucy2f. Visual function was assessed with an optomotor assay on 6-days-post- fertilization larvae, and by analyzing changes in retinal histology. Gucy2f knockdown resulted in significantly lower vision as measured by the optomotor response compared with uninjected and control MO-injected zebrafish larvae. Histological changes in the Gucy2f-knockdown larvae included loss and shortening of cone and rod outer segments. A zebrafish model of Gucy2f- related LCA1 displays early visual dysfunction and photoreceptor layer dystrophy. -

Autocrine IFN Signaling Inducing Profibrotic Fibroblast Responses By
Downloaded from http://www.jimmunol.org/ by guest on September 23, 2021 Inducing is online at: average * The Journal of Immunology , 11 of which you can access for free at: 2013; 191:2956-2966; Prepublished online 16 from submission to initial decision 4 weeks from acceptance to publication August 2013; doi: 10.4049/jimmunol.1300376 http://www.jimmunol.org/content/191/6/2956 A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Autocrine IFN Signaling Feng Fang, Kohtaro Ooka, Xiaoyong Sun, Ruchi Shah, Swati Bhattacharyya, Jun Wei and John Varga J Immunol cites 49 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2013/08/20/jimmunol.130037 6.DC1 This article http://www.jimmunol.org/content/191/6/2956.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2013 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 23, 2021. The Journal of Immunology A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Inducing Autocrine IFN Signaling Feng Fang,* Kohtaro Ooka,* Xiaoyong Sun,† Ruchi Shah,* Swati Bhattacharyya,* Jun Wei,* and John Varga* Activation of TLR3 by exogenous microbial ligands or endogenous injury-associated ligands leads to production of type I IFN. -

Supplemental Figures 04 12 2017
Jung et al. 1 SUPPLEMENTAL FIGURES 2 3 Supplemental Figure 1. Clinical relevance of natural product methyltransferases (NPMTs) in brain disorders. (A) 4 Table summarizing characteristics of 11 NPMTs using data derived from the TCGA GBM and Rembrandt datasets for 5 relative expression levels and survival. In addition, published studies of the 11 NPMTs are summarized. (B) The 1 Jung et al. 6 expression levels of 10 NPMTs in glioblastoma versus non‐tumor brain are displayed in a heatmap, ranked by 7 significance and expression levels. *, p<0.05; **, p<0.01; ***, p<0.001. 8 2 Jung et al. 9 10 Supplemental Figure 2. Anatomical distribution of methyltransferase and metabolic signatures within 11 glioblastomas. The Ivy GAP dataset was downloaded and interrogated by histological structure for NNMT, NAMPT, 12 DNMT mRNA expression and selected gene expression signatures. The results are displayed on a heatmap. The 13 sample size of each histological region as indicated on the figure. 14 3 Jung et al. 15 16 Supplemental Figure 3. Altered expression of nicotinamide and nicotinate metabolism‐related enzymes in 17 glioblastoma. (A) Heatmap (fold change of expression) of whole 25 enzymes in the KEGG nicotinate and 18 nicotinamide metabolism gene set were analyzed in indicated glioblastoma expression datasets with Oncomine. 4 Jung et al. 19 Color bar intensity indicates percentile of fold change in glioblastoma relative to normal brain. (B) Nicotinamide and 20 nicotinate and methionine salvage pathways are displayed with the relative expression levels in glioblastoma 21 specimens in the TCGA GBM dataset indicated. 22 5 Jung et al. 23 24 Supplementary Figure 4. -

Mrna Expression in Human Leiomyoma and Eker Rats As Measured by Microarray Analysis
Table 3S: mRNA Expression in Human Leiomyoma and Eker Rats as Measured by Microarray Analysis Human_avg Rat_avg_ PENG_ Entrez. Human_ log2_ log2_ RAPAMYCIN Gene.Symbol Gene.ID Gene Description avg_tstat Human_FDR foldChange Rat_avg_tstat Rat_FDR foldChange _DN A1BG 1 alpha-1-B glycoprotein 4.982 9.52E-05 0.68 -0.8346 0.4639 -0.38 A1CF 29974 APOBEC1 complementation factor -0.08024 0.9541 -0.02 0.9141 0.421 0.10 A2BP1 54715 ataxin 2-binding protein 1 2.811 0.01093 0.65 0.07114 0.954 -0.01 A2LD1 87769 AIG2-like domain 1 -0.3033 0.8056 -0.09 -3.365 0.005704 -0.42 A2M 2 alpha-2-macroglobulin -0.8113 0.4691 -0.03 6.02 0 1.75 A4GALT 53947 alpha 1,4-galactosyltransferase 0.4383 0.7128 0.11 6.304 0 2.30 AACS 65985 acetoacetyl-CoA synthetase 0.3595 0.7664 0.03 3.534 0.00388 0.38 AADAC 13 arylacetamide deacetylase (esterase) 0.569 0.6216 0.16 0.005588 0.9968 0.00 AADAT 51166 aminoadipate aminotransferase -0.9577 0.3876 -0.11 0.8123 0.4752 0.24 AAK1 22848 AP2 associated kinase 1 -1.261 0.2505 -0.25 0.8232 0.4689 0.12 AAMP 14 angio-associated, migratory cell protein 0.873 0.4351 0.07 1.656 0.1476 0.06 AANAT 15 arylalkylamine N-acetyltransferase -0.3998 0.7394 -0.08 0.8486 0.456 0.18 AARS 16 alanyl-tRNA synthetase 5.517 0 0.34 8.616 0 0.69 AARS2 57505 alanyl-tRNA synthetase 2, mitochondrial (putative) 1.701 0.1158 0.35 0.5011 0.6622 0.07 AARSD1 80755 alanyl-tRNA synthetase domain containing 1 4.403 9.52E-05 0.52 1.279 0.2609 0.13 AASDH 132949 aminoadipate-semialdehyde dehydrogenase -0.8921 0.4247 -0.12 -2.564 0.02993 -0.32 AASDHPPT 60496 aminoadipate-semialdehyde -

Sons of Martha: Reshaping the Electric Industry
A Novel GCAPl Missense Mutation (L151F) in a Large Family with Autosomal Dominant Cone-Rod Dystrophy (adCORD) Izabela Sokal,1 William J. Dupps,2 Michael A. Grassi,2 Jeremiah Bro wn, Jr,2 3 Louisa M. A ffatigato2 Nirmalya RoychowdhutyLili Yang,' Sfawomir Filipek,5 Krzysztof Palczewski,1 67 Edwin M. Stone,28 and Wolfgang Baehr'310 Purpose. To elucidate the phenotypic and biochemical charac ological [Ca2 f], consistent with a lowered affinity for Ca2 f- teristics of a novel mutation associated with autosomal domi binding to EF4. nant cone-rod dystrophy (adCORD). Conclusions. A novel L151F mutation in the EF4 hand domain Methods. Twenty-three family members of a CORD pedigree of GCAPl is associated with adCORD. The clinical phenotype underwent clinical examinations, including visual acuity tests, is characterized by early cone dysfunction and a progressive standardized full-field ERG, and fundus photography. Genomic loss of rod function. The biochemical phenotype is best de DNA was screened for mutations in GCAPl exons using DNA scribed as persistent stimulation of photoreceptor guanylate sequencing and single-strand conformational polymorphism cyclase, representing a gain of function of mutant GCAPl. (SSCP) analysis. Function and stability of recombinant GCAP1- Although a conservative substitution, molecular dynamics sug L151F were tested as a function of [Ca2 f], and its structure was gests a significant change in Ca2 ^-binding to EF4 and EF2 and changes in the shape of L151F-GCAP1. {Invest Ophthalmol Vis probed by molecular dynamics. Sci. 2005;46:1124-1132) DOklO.l l67/iovs.04-1431 Results. Affected family members experienced dyschromatop- sia, hemeralopia, and reduced visual acuity by the second to third decade of life.