A Dissertation Entitled Development of Selective Inhibitors Against
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Namely, Aspartokinase and Aspartate 3-Semialdehyde Dehydrogenase) Are Not Subject to Feedback Inhibition Control by the End Product Methionine
REGULATION OF HOMOSERINE BIOSYNTHESIS BY L-CYSTEINE, A TERMINA L METABOLITE OF A LINKED PA THWA Y* By PRASANTA DATTA DEPARTMENT OF BIOLOGICAL CHEMISTRY, UNIVERSITY OF MICHIGAN, ANN ARBOR Communicated by Martin D. Kamen, June 23, 1967 In bacteria, the amino acid homoserine is a key branch-point intermediate in the synthesis of several amino acids of the aspartic pathway.' On the one hand, homoserine is converted to threonine (and thus to isoleucine), and through a separate sequence of reactions is transformed to methionine.1 In the latter se- quence, a succinylated product of homoserine is condensed with cysteine to pro- duce cystathionine,2-5 a precursor of methionine. Recent studies (see refs. 6 and 7 for up-to-date reviews on the subject) with several microorganisms have revealed that the synthesis of homoserine is regulated by various combinations of repression and/or feedback inhibition controls of early biosynthetic enzymes of the aspartic pathway by several end-product metabolites. One of these enzymes, homoserine dehydrogenase, catalyzes the pyridine nucleotide-linked reduction of aspartate j3-semialdehyde to homoserine. In the various bacteria that have been examined, this dehydrogenase as well as the two earlier enzymes of the path- way (namely, aspartokinase and aspartate 3-semialdehyde dehydrogenase) are not subject to feedback inhibition control by the end product methionine. This report describes the results of experiments on the control of activity of several bacterial homoserine dehydrogenases by L-cysteine, a terminal metabolite of a linked or connecting pathway, required for the synthesis of cystathionine.2-5 The findings suggest that the size of the homoserine pool in bacterial cells must depend, at least in part, on complex interdependent regulatory interactions of both cysteine and threonine on homoserine dehydrogenase. -
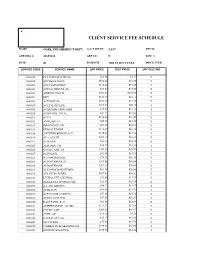
Oakland Sheriff's Dept Fee Schedule 02.09
CLIENT SERVICE FEE SCHEDULE NAME: OAKLAND SHERIFF'S DEPT ACCT SETUP: 2/1/87 REP ID: GRP BILL #: 22857610 GRP CD: N DISC %: FS ID: 01 FS DESCR: 2003 CLIENT FEES PRICE TYPE: SERVICE CODE SERVICE NAME UPF PRICE TEST PRICE UPF DISC IND 0000018 PLT SODIUM CITRATE $12.86 $4.37 Y 0000022 APC RESISTANCE $101.62 $34.55 Y 0000201 ACETAMINOPHEN $110.24 $37.48 Y 0000202 ACETALDEHYDE (B) $73.00 $73.00 N 0000203 ARSENIC,TISSUE $302.00 $302.00 N 0000204 DHT $182.70 $62.12 Y 0000205 ACETONE (B) $102.20 $34.75 Y 0000206 ACETYLCHOLINE $165.00 $56.10 Y 0000208 ACID PHOS, PROS, IMM $35.60 $12.10 Y 0000210 ACID PHOS, TOTAL $37.22 $12.65 Y 0000211 ACTH $110.00 $37.40 Y 0000212 AMYLASE (U) $45.45 $15.45 Y 0000213 IMMUNOFIX, UR $67.39 $22.91 Y 0000214 ETHOSUXIMIDE $112.07 $38.10 Y 0000216 ANTITHROMBIN III ACT $170.46 $57.96 Y 0000219 ALA, QUANT $107.10 $36.41 Y 0000223 ALBUMIN $22.88 $22.88 N 0000224 ALBUMIN, CSF $36.25 $12.33 Y 0000225 CYCLIC AMP, UR $205.80 $69.97 Y 0000227 ALDOLASE $32.08 $10.91 Y 0000228 A-2-MACROGLOB. $78.75 $26.78 Y 0000229 ALDOSTERONE (U) $251.58 $85.54 Y 0000230 ALDOSTERONE $207.10 $70.41 Y 0000231 ALK PHOS ISOENZYMES $61.34 $20.86 Y 0000232 AFP, FLUID W/RFX $107.40 $36.52 Y 0000233 LEUKOCYTE ALK PHOS $34.60 $11.76 Y 0000234 ALKALINE PHOSPHATASE $22.88 $22.88 N 0000235 A-1-ANTITRYPSN $40.22 $13.67 Y 0000236 AMIKACIN $99.91 $33.97 Y 0000237 AFP,TUMOR (CHIRON) $53.86 $18.31 Y 0000238 AMINO ACID SCR $87.15 $29.63 Y 0000240 RAST PANEL #103 $62.00 $62.00 N 0000241 AMPHETAMINE GC/MS $111.70 $37.98 Y 0000242 CYCLIC AMP $205.80 $69.97 Y 0000243 AMYLASE $16.42 $5.58 Y 0000248 HACKBERRY IGE $35.40 $35.40 N 0000249 ANA W/RFX $55.00 $18.70 Y 0000250 *AMIKACIN,PEAK&TROUGH $199.83 $67.94 Y 0000251 ANDROSTENEDIONE $180.65 $61.42 Y 0000252 ANTI-DIUR. -

Joseph M. Jez • Department of Biology • Washington University in St
Joseph M. Jez • Department of Biology • Washington University in St. Louis Joseph M. Jez Professor and Howard Hughes Medical Institute Professor Department of Biology, Washington University in St. Louis One Brookings Drive, Campus Box 1137, St. Louis, MO 63130-4899 Phone: 314-935-3376; Email: [email protected] –––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––– EDUCATION University of Pennsylvania, Philadelphia, PA Ph.D. Biochemistry & Molecular Biophysics, 1998 Thesis: Steroid Recognition and Engineering of Catalysis in Mammalian Aldo-Keto Reductases Research Advisor: Prof. Trevor M. Penning Penn State University, University Park, PA B.S. Biochemistry (with Honors and English minor), 1992 Research Advisor: Prof. Gregory K. Farber PROFESSIONAL EXPERIENCE Washington University in St. Louis, St. Louis, MO Professor, Department of Biology (2015-current) Co-Director, Plant and Microbial Biosciences Program, Division of Biology & Biomedical Sciences (2013-current) Associate Professor, Department of Biology (2011-2015) Assistant Professor, Department of Biology (2008-2011) Honorary Assistant Professor, Department of Biology (2006-2008) Donald Danforth Plant Science Center, St. Louis, MO Assistant Member & Principal Investigator (2002-2010) Kosan Biosciences, Hayward, CA Scientist, New Technology Group (2001-2002) Supervisor: Dr. Daniel V. Santi The Salk Institute for Biological Studies, La Jolla, CA NIH-NRSA Postdoctoral Research Fellow, Structural Biology Laboratory (1998-2001) Project: Structure, Mechanism, -

Yeast Genome Gazetteer P35-65
gazetteer Metabolism 35 tRNA modification mitochondrial transport amino-acid metabolism other tRNA-transcription activities vesicular transport (Golgi network, etc.) nitrogen and sulphur metabolism mRNA synthesis peroxisomal transport nucleotide metabolism mRNA processing (splicing) vacuolar transport phosphate metabolism mRNA processing (5’-end, 3’-end processing extracellular transport carbohydrate metabolism and mRNA degradation) cellular import lipid, fatty-acid and sterol metabolism other mRNA-transcription activities other intracellular-transport activities biosynthesis of vitamins, cofactors and RNA transport prosthetic groups other transcription activities Cellular organization and biogenesis 54 ionic homeostasis organization and biogenesis of cell wall and Protein synthesis 48 plasma membrane Energy 40 ribosomal proteins organization and biogenesis of glycolysis translation (initiation,elongation and cytoskeleton gluconeogenesis termination) organization and biogenesis of endoplasmic pentose-phosphate pathway translational control reticulum and Golgi tricarboxylic-acid pathway tRNA synthetases organization and biogenesis of chromosome respiration other protein-synthesis activities structure fermentation mitochondrial organization and biogenesis metabolism of energy reserves (glycogen Protein destination 49 peroxisomal organization and biogenesis and trehalose) protein folding and stabilization endosomal organization and biogenesis other energy-generation activities protein targeting, sorting and translocation vacuolar and lysosomal -

Endophytic Fungi: Treasure for Anti-Cancerous Compounds
International Journal of Pharmacy and Pharmaceutical Sciences ISSN- 0975-1491 Vol 8, Issue 8, 2016 Review Article ENDOPHYTIC FUNGI: TREASURE FOR ANTI-CANCEROUS COMPOUNDS ANAND DILIP FIRODIYAa*, RAJESH KUMAR TENGURIAb aCSRD, Peoples University, Bhopal 462037, Madhya Pradesh, India, bDepartment of Botany, Govt. PG College, Rajgarh 496551, Madhya Pradesh, India Email: [email protected] Received: 22 Apr 2016 Revised and Accepted: 20 June 2016 ABSTRACT Endophytic fungi that live asymptomatically inside the plant tissues have novel bioactive metabolites exhibiting a variety of biological activities, especially against cancer. This review highlights the research progress on the production of anticancer compounds by endophytic fungi from 1990- 2015. Anticancer activity is generally associated with the cytotoxicity of the compounds present in the endophytic fungi. The ubiquitous nature of endophytic fungi synthesise diverse chemicals with promising anticancer activity from either their original host or related species. Modification in fermentation parameters and genetic insight of endophytes may produce novel anti-cancerous compounds. Keywords: Cancer, Medicinal plants, Secondary metabolites © 2016 The Authors. Published by Innovare Academic Sciences Pvt Ltd. This is an open access article under the CC BY license (http://creativecommons.org/licenses/by/4.0/) INTRODUCTION endophytic fungi detectable by high-performance liquid chromate- graphy, nuclear magnetic resonance, mass spectrophotometer and The interest in the biogenic medicines has revived throughout the X-ray crystallography and its cytotoxicity of the bioactive world, as the increase in awareness of the health hazards and compounds against cancer cell lines. The compounds with potential toxicity associated with the random use of synthetic drugs and application were also considered in the selection of antitumor antibiotics [1]. -

Table S1. List of Genes Up- Or Down-Regulated in H99 When Bound by 18B7
Table S1. List of genes up- or down-regulated in H99 when bound by 18B7. Up-regulated Genes Classification CNF03470: Formate dehydrogenase Metabolism CNC03960: Phosphate transporter, putative Metabolism CND00530: Putative urea transporter Secretion CNA02250: Ammonium transporter MEP1 Secretion CNC06440: Inositol 1-phosphate synthase Metabolism CND03490: Chitin deacetylase-like mannoprotein MP98 Cell wall CNA04560: Hypothetical protein Hypothetical CNF02180: Acetyl-CoA carboxylase, putative Metabolism CNJ00690: Uracil permease, putative Secretion CNF02510: Alcohol dehydrogenase, putative Metabolism CNE04360: Fatty acid synthase, alpha subunit-related Metabolism CNM00180: Cyclohydrolase, putative Metabolism CNL03740: AF540951 catalase isozyme P Stress CNE04370: Fatty acid synthase, beta subunit Metabolism CNA01790: Expressed protein Hypothetical CNA05700: Expressed protein Hypothetical CND03840: Vacuole fusion, non-autophagic-related protein, putative Cell wall CNM00980: Hypothetical protein Hypothetical CNI02420: Uricase (Urate oxidase), putative Metabolism CNJ01090: Xylitol dehydrogenase-related Cell wall CNC03430: Alpha-1,6-mannosyltransferase, putative Cell wall CNF04120: Expressed protein Hypothetical CNB01810: Short chain dehydrogenase, putative Metabolism CNJ01200: Hypothetical protein Hypothetical CNA01160: LSDR putative Metabolism CNH03430: Hypothetical protein Hypothetical CNM02410: Putative proteine disulfate isomerase Housekeeping CNG04200: Alpha-amylase, putative Cell wall CNC00920: NADP-dependent glutamate dehydrogenase Metabolism -

Supplemental Methods
Supplemental Methods: Sample Collection Duplicate surface samples were collected from the Amazon River plume aboard the R/V Knorr in June 2010 (4 52.71’N, 51 21.59’W) during a period of high river discharge. The collection site (Station 10, 4° 52.71’N, 51° 21.59’W; S = 21.0; T = 29.6°C), located ~ 500 Km to the north of the Amazon River mouth, was characterized by the presence of coastal diatoms in the top 8 m of the water column. Sampling was conducted between 0700 and 0900 local time by gently impeller pumping (modified Rule 1800 submersible sump pump) surface water through 10 m of tygon tubing (3 cm) to the ship's deck where it then flowed through a 156 µm mesh into 20 L carboys. In the lab, cells were partitioned into two size fractions by sequential filtration (using a Masterflex peristaltic pump) of the pre-filtered seawater through a 2.0 µm pore-size, 142 mm diameter polycarbonate (PCTE) membrane filter (Sterlitech Corporation, Kent, CWA) and a 0.22 µm pore-size, 142 mm diameter Supor membrane filter (Pall, Port Washington, NY). Metagenomic and non-selective metatranscriptomic analyses were conducted on both pore-size filters; poly(A)-selected (eukaryote-dominated) metatranscriptomic analyses were conducted only on the larger pore-size filter (2.0 µm pore-size). All filters were immediately submerged in RNAlater (Applied Biosystems, Austin, TX) in sterile 50 mL conical tubes, incubated at room temperature overnight and then stored at -80oC until extraction. Filtration and stabilization of each sample was completed within 30 min of water collection. -

And Mushroom-Associated Alkaloids from Two Behavior Modifying
bioRxiv preprint doi: https://doi.org/10.1101/375105; this version posted December 18, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Psychoactive plant- and mushroom-associated alkaloids from two behavior modifying 2 cicada pathogens 3 4 Greg R. Boyce1, Emile Gluck-Thaler2, Jason C. Slot2, Jason E. Stajich3, William J. Davis4, Tim 5 Y. James4, John R. Cooley5, Daniel G. Panaccione1, Jørgen Eilenberg6, Henrik H. De Fine Licht6, 6 Angie M. Macias1, Matthew C. Berger1, Kristen L. Wickert1, Cameron M. Stauder1, Ellie J. 7 Spahr1, Matthew D. Maust1, Amy M. Metheny1, Chris Simon5, Gene Kritsky7, Kathie T. Hodge8, 8 Richard A. Humber8,9, Terry Gullion10, Dylan P. G. Short11, Teiya Kijimoto1, Dan Mozgai12, 9 Nidia Arguedas13, Matt T. Kasson1,* 10 11 1Division of Plant and Soil Sciences, West Virginia University, Morgantown, West Virginia, 12 26506, USA. 13 2Department of Plant Pathology, The Ohio State University, Columbus, Ohio 43210, USA. 14 3Department of Microbiology and Plant Pathology and Institute for Integrative Genome Biology, 15 University of California, Riverside, California 92521, USA. 16 4Department of Ecology and Evolution, University of Michigan, Ann Arbor, Michigan 48109, 17 USA. 18 5Department of Ecology and Evolutionary Biology, University of Connecticut, Storrs, 19 Connecticut 06269, USA. 20 6Department of Plant and Environmental Science, University of Copenhagen, Denmark. 21 7Department of Biology, Mount St. Joseph University, Cincinnati, Ohio 45233, USA. 22 8Plant Pathology & Plant-Microbe Biology, School of Integrative Plant Science, Cornell 23 University, Ithaca, New York 14853, USA. -
Fungal Endophytes As Efficient Sources of Plant-Derived Bioactive
microorganisms Review Fungal Endophytes as Efficient Sources of Plant-Derived Bioactive Compounds and Their Prospective Applications in Natural Product Drug Discovery: Insights, Avenues, and Challenges Archana Singh 1,2, Dheeraj K. Singh 3,* , Ravindra N. Kharwar 2,* , James F. White 4,* and Surendra K. Gond 1,* 1 Department of Botany, MMV, Banaras Hindu University, Varanasi 221005, India; [email protected] 2 Department of Botany, Institute of Science, Banaras Hindu University, Varanasi 221005, India 3 Department of Botany, Harish Chandra Post Graduate College, Varanasi 221001, India 4 Department of Plant Biology, Rutgers University, New Brunswick, NJ 08901, USA * Correspondence: [email protected] (D.K.S.); [email protected] (R.N.K.); [email protected] (J.F.W.); [email protected] (S.K.G.) Abstract: Fungal endophytes are well-established sources of biologically active natural compounds with many producing pharmacologically valuable specific plant-derived products. This review details typical plant-derived medicinal compounds of several classes, including alkaloids, coumarins, flavonoids, glycosides, lignans, phenylpropanoids, quinones, saponins, terpenoids, and xanthones that are produced by endophytic fungi. This review covers the studies carried out since the first report of taxol biosynthesis by endophytic Taxomyces andreanae in 1993 up to mid-2020. The article also highlights the prospects of endophyte-dependent biosynthesis of such plant-derived pharma- cologically active compounds and the bottlenecks in the commercialization of this novel approach Citation: Singh, A.; Singh, D.K.; Kharwar, R.N.; White, J.F.; Gond, S.K. in the area of drug discovery. After recent updates in the field of ‘omics’ and ‘one strain many Fungal Endophytes as Efficient compounds’ (OSMAC) approach, fungal endophytes have emerged as strong unconventional source Sources of Plant-Derived Bioactive of such prized products. -

Construction of a Synthetic Metabolic Pathway for the Production of 2,4
Construction of a synthetic metabolic pathway for the production of 2,4-dihydroxybutyric acid from homoserine Thomas Walther, Florence Calvayrac, Yoann Malbert, Ceren Alkim, Clémentine Dressaire, Hélène Cordier, Jean Marie François To cite this version: Thomas Walther, Florence Calvayrac, Yoann Malbert, Ceren Alkim, Clémentine Dressaire, et al.. Construction of a synthetic metabolic pathway for the production of 2,4-dihydroxybutyric acid from homoserine. Metabolic Engineering, Elsevier, 2018, 10.1016/j.ymben.2017.12.005. hal-01886438 HAL Id: hal-01886438 https://hal.archives-ouvertes.fr/hal-01886438 Submitted on 27 May 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution| 4.0 International License Author’s Accepted Manuscript Construction of a synthetic metabolic pathway for the production of 2,4-dihydroxybutyric acid from homoserine Thomas Walther, Florence Calvayrac, Yoann Malbert, Ceren Alkim, Clémentine Dressaire, Hélène Cordier, Jean Marie François www.elsevier.com/locate/ymben PII: S1096-7176(17)30350-6 -

Paraburkholderia Phymatum Homocitrate Synthase Nifv Plays a Key Role for Nitrogenase Activity During Symbiosis with Papilionoids and in Free-Living Growth Conditions
cells Article Paraburkholderia phymatum Homocitrate Synthase NifV Plays a Key Role for Nitrogenase Activity during Symbiosis with Papilionoids and in Free-Living Growth Conditions Paula Bellés-Sancho 1 , Martina Lardi 1, Yilei Liu 1, Sebastian Hug 1, Marta Adriana Pinto-Carbó 1, Nicola Zamboni 2 and Gabriella Pessi 1,* 1 Department of Plant and Microbial Biology, University of Zürich, CH-8057 Zürich, Switzerland; [email protected] (P.B.-S.); [email protected] (M.L.); [email protected] (Y.L.); [email protected] (S.H.); [email protected] (M.A.P.-C.) 2 ETH Zürich, Institute of Molecular Systems Biology, CH-8093 Zürich, Switzerland; [email protected] * Correspondence: [email protected]; Tel.: +41-44-63-52904 Abstract: Homocitrate is an essential component of the iron-molybdenum cofactor of nitrogenase, the bacterial enzyme that catalyzes the reduction of dinitrogen (N2) to ammonia. In nitrogen-fixing and nodulating alpha-rhizobia, homocitrate is usually provided to bacteroids in root nodules by their plant host. In contrast, non-nodulating free-living diazotrophs encode the homocitrate synthase (NifV) and reduce N2 in nitrogen-limiting free-living conditions. Paraburkholderia phymatum STM815 is a beta-rhizobial strain, which can enter symbiosis with a broad range of legumes, including Citation: Bellés-Sancho, P.; Lardi, M.; papilionoids and mimosoids. In contrast to most alpha-rhizobia, which lack nifV, P. phymatum Liu, Y.; Hug, S.; Pinto-Carbó, M.A.; harbors a copy of nifV on its symbiotic plasmid. We show here that P. phymatum nifV is essential for Zamboni, N.; Pessi, G. -

Identification of Six Novel Allosteric Effectors of Arabidopsis Thaliana Aspartate Kinase-Homoserine Dehydrogenase
Identification of six novel allosteric effectors of Arabidopsis thaliana Aspartate kinase-Homoserine dehydrogenase. Physiological context sets the specificity Gilles Curien, Stéphane Ravanel, Mylène Robert, Renaud Dumas To cite this version: Gilles Curien, Stéphane Ravanel, Mylène Robert, Renaud Dumas. Identification of six novel allosteric effectors of Arabidopsis thaliana Aspartate kinase-Homoserine dehydrogenase. Physiological context sets the specificity. Journal of Biological Chemistry, American Society for Biochemistry and Molecular Biology, 2005, 280 (50), pp.41178-41183. 10.1074/jbc.M509324200. hal-00016689 HAL Id: hal-00016689 https://hal.archives-ouvertes.fr/hal-00016689 Submitted on 31 May 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 280, NO. 50, pp. 41178–41183, December 16, 2005 © 2005 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in the U.S.A. Identification of Six Novel Allosteric Effectors of Arabidopsis thaliana Aspartate Kinase-Homoserine Dehydrogenase