Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Nitrogen Oxide - Wikipedia, the Free Encyclopedia Page 1 of 3
Nitrogen oxide - Wikipedia, the free encyclopedia Page 1 of 3 Nitrogen oxide From Wikipedia, the free encyclopedia Nitrogen oxide can refer to a binary compound of oxygen and nitrogen, or a mixture of such compounds: Contents ■ Nitric oxide (NO), nitrogen(II) oxide ■ Nitrogen dioxide (NO2), nitrogen(IV) oxide ■ 1 NOx ■ Nitrous oxide (N2O), nitrogen(I) oxide ■ 2 Derivatives ■ Nitrate radical (NO3), nitrogen(VI) oxide ■ 3 See also ■ Dinitrogen trioxide (N2O3), nitrogen(II,IV) oxide ■ 4 References ■ Dinitrogen tetroxide (N2O4), nitrogen(IV) oxide ■ Dinitrogen pentoxide (N2O5), nitrogen(V) oxide In atmospheric chemistry and air pollution and related fields, nitrogen oxides refers specifically to NOx [1][2] (NO and NO2). Only the first three of these compounds can be isolated at room temperature. N2O3, N2O4, and N2O5 all decompose rapidly at room temperature. Nitrate radical is very reactive. N2O is stable and rather unreactive at room temperature, while NO and NO2 are quite reactive but nevertheless quite stable when isolated. Dinitrogen trioxide, Nitric oxide, NO Nitrogen dioxide, NO2 Nitrous oxide, N2O N2O3 Dinitrogen tetroxide, Dinitrogen pentoxide, N2O4 N2O5 http://en.wikipedia.org/wiki/Nitrogen_oxide 11/2/2010 Nitrogen oxide - Wikipedia, the free encyclopedia Page 2 of 3 NOx Main article: NOx NOx (often written NOx) refers to NO and NO2. They are produced during combustion, especially at high temperature. These two chemicals are important trace species in Earth's atmosphere. In the troposphere, during daylight, NO reacts with partly oxidized organic species (or the peroxy radical) to form NO2, which is then photolyzed by sunlight to reform NO: NO + CH3O2 → NO2 + CH3O NO2 + sunlight → NO + O The oxygen atom formed in the second reaction then goes on to form ozone; this series of reactions is the main source of tropospheric ozone. -

COVALENT COMPOUNDS Unit 2 COVALENT BOND
COVALENT COMPOUNDS Unit 2 COVALENT BOND Forms between two non-metals! Electrons are shared! Co= share Valent = electrons Since electrons are shared, no need to balance charges Covalent bonds form molecules. Molecule - group of neutral atoms held together with a covalent bond Diatomic molecules – molecule consisting of 2 atoms MOLECULES H2O2Br2F2I2N2Cl2 Compound composed of molecules is a molecular compound. MOLECULAR COMPOUNDS Molecular Compounds Properties Gases or liquids Low melting/boiling points Usually made from 2 nonmetals bonded covalently Do not conduct electricity (solid or dissolved) Molecular chemical formula shows how many atoms of each element a molecule contains. H2O – Molecular formula of water 2 Hydrogen atoms, 1 Oxygen atom CO2 – Molecular formula for Carbon Dioxide MOLECULAR 1 Carbon atom, 2 Oxygen atoms FORMULAS H:H - Lewis Dot H-H – Structural Formula H2 – Molecular chemical formula LEWIS STRUCTURES COVALENT BONDING To draw a Lewis Structure, one must know the types of atoms in the molecule, the number of atoms in the molecule and the number of valence electrons for each atom in the molecule. DOUBLE & TRIPLE COVALENT BONDS Sharing a pair of electrons creates a single covalent bond Sharing more than one pair of electrons creates double and triple covalent bonds. 2 shared pairs – Double covalent bond 3 shared pairs – Triple covalent bond Carbon, Nitrogen, Oxygen 1. First element is named first, using the full name of the element 2. The second element is named by dropping ending and adding RULES FOR “–ide” NAMING 3. Prefixes are used to denote # COVALENT of each atom present COMPOUNDS 4. Prefix mono- is never used on the first element 5. -
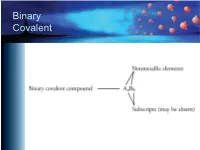
Binary Covalent Common Names
Binary Covalent Common Names – H2O, water – NH3, ammonia – CH4, methane – C2H6, ethane – C3H8, propane – C4H10, butane – C5H12, pentane – C6H14, hexane Naming Binary Covalent CompounDs • If the subscript for the first element is greater than one, inDicate the subscript with a prefix. – We Do not write mono- on the first name. – Leave the "a" off the enD of the prefixes that enD in "a" anD the “o” off of mono- if they are placeD in front of an element that begins with a vowel (oxygen or ioDine). Prefixes mon(o) hex(a) di hept(a) tri oct(a) tetr(a) non(a) pent(a) dec(a) Nitrogen OxiDe Names • N2O3 – name starts with di • N2O5 – name starts with di • NO2 – no initial prefix • NO – no initial prefix Naming Binary Covalent CompounDs • Follow the prefix with the name of the first element in the formula. – N2O3 – dinitrogen – N2O3 – dinitrogen – NO2 – nitrogen – NO – nitrogen Naming Binary Covalent CompounDs • Write a prefix to inDicate the subscript for the seconD element. (Remember to leave the “o” off of mono- anD the “a” off of the prefixes that enD in “a” when they are placeD in front of a name that begins with a vowel.) – N2O3 – dinitrogen tri – N2O5 – dinitrogen pent – NO2 – nitrogen di – NO – nitrogen mon Naming Binary Covalent CompounDs • Write the root of the name of the seconD symbol in the formula. (See the next sliDe.) – N2O3 – dinitrogen triox – N2O5 – dinitrogen pentox – NO2 – nitrogen diox – NO – nitrogen monox Roots of Nonmetals H hydr- F fluor- C carb- Cl chlor- N nitr- Br brom- P phosph- I iod- O ox- S sulf- Se selen- Naming Binary Covalent CompounDs • AdD -ide to the enD of the name. -

Nitrate Formation from Heterogeneous Uptake of Dinitrogen Pentoxide During a Severe Winter Haze in Southern China” by Hui Yun Et Al
Atmos. Chem. Phys. Discuss., https://doi.org/10.5194/acp-2018-698-RC1, 2018 © Author(s) 2018. This work is distributed under the Creative Commons Attribution 4.0 License. Interactive comment on “Nitrate formation from heterogeneous uptake of dinitrogen pentoxide during a severe winter haze in southern China” by Hui Yun et al. Anonymous Referee #1 Received and published: 23 September 2018 The manuscript of Yun et al., reported half month measurement of N2O5, ClNO2 and other relative parameters during heavy haze episodes in Pearl River Delta (PRD) of southern China. The N2O5 uptake coefficient and ClNO2 yield were determined from the observations. The study showed the observation evidence of the enhancement of particulate nitrate in the first several hours can be fully explained by the N2O5 het- erogeneous hydrolysis and even comparable with the nitric acid formed by OH+NO2 during daytime. Overall, the paper contributes to the knowledge of N2O5 heteroge- neous chemistry and highlight the heterogeneous reactions in the formation of partic- ulate nitrate in southern China. The following comments should be addressed before publishing on ACP. C1 Major comments: The steady state assumption to derive the N2O5 uptake coefficient needs to be verified by model simulations under the observed conditions (with input from NO, NO2, O3, VOCs). It is useful to try other method (e.g. Brown et al., 2006) to derive N2O5 uptake coefficient. Brown, S. S., Ryerson, T. B., Wollny, A. G., Brock, C. A., Peltier, R., Sullivan, A. P., Weber, R. J., Dube, W. P., Trainer, M., Meagher, J. F., Fehsenfeld, F. C., and Ravishankara, A. -

Chemical Names and CAS Numbers Final
Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number C3H8O 1‐propanol C4H7BrO2 2‐bromobutyric acid 80‐58‐0 GeH3COOH 2‐germaacetic acid C4H10 2‐methylpropane 75‐28‐5 C3H8O 2‐propanol 67‐63‐0 C6H10O3 4‐acetylbutyric acid 448671 C4H7BrO2 4‐bromobutyric acid 2623‐87‐2 CH3CHO acetaldehyde CH3CONH2 acetamide C8H9NO2 acetaminophen 103‐90‐2 − C2H3O2 acetate ion − CH3COO acetate ion C2H4O2 acetic acid 64‐19‐7 CH3COOH acetic acid (CH3)2CO acetone CH3COCl acetyl chloride C2H2 acetylene 74‐86‐2 HCCH acetylene C9H8O4 acetylsalicylic acid 50‐78‐2 H2C(CH)CN acrylonitrile C3H7NO2 Ala C3H7NO2 alanine 56‐41‐7 NaAlSi3O3 albite AlSb aluminium antimonide 25152‐52‐7 AlAs aluminium arsenide 22831‐42‐1 AlBO2 aluminium borate 61279‐70‐7 AlBO aluminium boron oxide 12041‐48‐4 AlBr3 aluminium bromide 7727‐15‐3 AlBr3•6H2O aluminium bromide hexahydrate 2149397 AlCl4Cs aluminium caesium tetrachloride 17992‐03‐9 AlCl3 aluminium chloride (anhydrous) 7446‐70‐0 AlCl3•6H2O aluminium chloride hexahydrate 7784‐13‐6 AlClO aluminium chloride oxide 13596‐11‐7 AlB2 aluminium diboride 12041‐50‐8 AlF2 aluminium difluoride 13569‐23‐8 AlF2O aluminium difluoride oxide 38344‐66‐0 AlB12 aluminium dodecaboride 12041‐54‐2 Al2F6 aluminium fluoride 17949‐86‐9 AlF3 aluminium fluoride 7784‐18‐1 Al(CHO2)3 aluminium formate 7360‐53‐4 1 of 75 Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number Al(OH)3 aluminium hydroxide 21645‐51‐2 Al2I6 aluminium iodide 18898‐35‐6 AlI3 aluminium iodide 7784‐23‐8 AlBr aluminium monobromide 22359‐97‐3 AlCl aluminium monochloride -

The Multiphase Chemical Kinetics of Dinitrogen Pentoxide with Aerosol Particles
The Multiphase Chemical Kinetics of Dinitrogen Pentoxide with Aerosol Particles Inauguraldissertation der Philosophisch-naturwissenschaftlichen Fakultät der Universität Bern vorgelegt von Goran Gržinić aus Kroatien/Italien Leiter der Arbeit: Prof. Dr. Andreas Türler Departement für Chemie und Biochemie The Multiphase Chemical Kinetics of Dinitrogen Pentoxide with Aerosol Particles Inauguraldissertation der Philosophisch-naturwissenschaftlichen Fakultät der Universität Bern vorgelegt von Goran Gržinić aus Kroatien/Italien Leiter der Arbeit: Prof. Dr. Andreas Türler Departement für Chemie und Biochemie Von der Philosophisch-naturwissenschaftlichen Fakultät angenommen. Bern, 16.12.2014 Der Dekan: Prof. Dr. Gilberto Colangelo It matters not how strait the gate, How charged with punishments the scroll, I am the master of my fate, I am the captain of my soul Invictus, William Ernest Henley Abstract Dinitrogen pentoxide (N2O5) is an important reactive intermediate in the nighttime chemistry of nitrogen oxides (NOx). As one of the major NOx sinks, it plays an important role in the regulation of the oxidative capacity of the atmosphere. 13 13 The N radioactive isotope tracer technique was used to synthesize N labeled N2O5 for the first time, and an experimental system for the study of uptake kinetics of N2O5 on aerosol particles was developed. A computer model of the gas phase chemistry involved was created to help optimizing the production of N2O5. The experimental 13 setup was successfully tested, and routine production of N2O5 in the ppb range was established. The uptake of N2O5 on citric acid aerosol was studied over an atmospherically relevant humidity range (17-70% RH). Citric acid was used as a proxy for highly oxidized organic species present in ambient organic aerosol. -

Nitrogen Oxides (Nox), Why and How They Are Controlled
United States Office of Air Quality EPA 456/F-99-006R Environmental Protection Planning and Standards November 1999 Agency Research Triangle Park, NC 27711 Air EPA-456/F-99-006R November 1999 Nitrogen Oxides (NOx), Why and How They Are Controlled Prepared by Clean Air Technology Center (MD-12) Information Transfer and Program Integration Division Office of Air Quality Planning and Standards U.S. Environmental Protection Agency Research Triangle Park, North Carolina 27711 DISCLAIMER This report has been reviewed by the Information Transfer and Program Integration Division of the Office of Air Quality Planning and Standards, U.S. Environmental Protection Agency and approved for publication. Approval does not signify that the contents of this report reflect the views and policies of the U.S. Environmental Protection Agency. Mention of trade names or commercial products is not intended to constitute endorsement or recommendation for use. Copies of this report are available form the National Technical Information Service, U.S. Department of Commerce, 5285 Port Royal Road, Springfield, Virginia 22161, telephone number (800) 553-6847. CORRECTION NOTICE This document, EPA-456/F-99-006a, corrects errors found in the original document, EPA-456/F-99-006. These corrections are: Page 8, fourth paragraph: “Destruction or Recovery Efficiency” has been changed to “Destruction or Removal Efficiency;” Page 10, Method 2. Reducing Residence Time: This section has been rewritten to correct for an ambiguity in the original text. Page 20, Table 4. Added Selective Non-Catalytic Reduction (SNCR) to the table and added acronyms for other technologies. Page 29, last paragraph: This paragraph has been rewritten to correct an error in stating the configuration of a typical cogeneration facility. -

Chemical Bonding
Chemical Bonding Part Two: Covalent Bonds Electronegativity and Bond Character v Electronegativity - the relative ability of an atom to attract electrons in a chemical bond vThe greater the electronegativity difference, the stronger the bond is that forms between them vIf the electronegativity difference is: • Less than 0.5, then the bond is nonpolar covalent • Between 0.5 and 1.6, the bond is polar covalent • Greater than 1.6, then the bond is ionic Calculating Bond Character Electronegativity Determine the type of bond: Chart Carbon 2.55 1. H and O 3.44-2.20 = 1.2 Nitrogen 3.04 POLAR COVALENT Potassium 0.82 2. Ca and F 3.98-1.00 = 2.98 Fluorine 3.98 IONIC Hydrogen 2.20 Oxygen 3.44 3. Ba and O 3.44-0.89 = 2.55 Calcium 1.00 IONIC Chlorine 3.98 4. C and O 3.44-2.55 = 0.89 Barium 0.89 POLAR COVALENT Bromine 2.96 5. N and N 0-NONPOLAR COVALENT Covalent Bonds vCharacterized by a sharing of electrons vThe attraction of two atoms for a shared pair of electrons is called a covalent bond v In a covalent bond, atoms share electrons and neither atom has an ionic charge vA compound whose atoms are held together by covalent bonds is a covalent compound vMolecules are formed from covalent bonds Single and Multiple Covalent Bonds v When more than one pair of electrons are shared between atoms multiple covalent bonds form vThese can be double or triple bonds depending on the number of pairs of electrons shared Single and Multiple Covalent Bonds Characteristics of Covalent Bonds v Low melting points v Do not conduct electricity in any state v Solids are often soft or brittle v Many are gases at room temperature v Most are less soluble in water than ionic compounds and are not electrolytes Polar vs. -

100 Most Important Chemical Compounds
64. Nitric Oxide 65. Nitrogen Dioxide 66. Nitrous Oxide CHEMICAL NAME = nitric monoxide nitrogen dioxide dinitrogen oxide CAS NUMBER = 10102–43–9 10102–44–0 10024–97–2 = MOLECULAR FORMULA NO NO2 N2O MOLAR MASS = 30.0 g/mol 46.0 g/mol 44.0 g/mol COMPOSITION = N(46.7%) O(53.3%) N(30.4%) O(69.6%) N(63.7%) MELTING POINT = −164°C −11.2 −90.8°C BOILING POINT = −152°C 21.1°C −88.6°C DENSITY = 1.35 g/L 2.05 g/L .99 g/L VAPOR DENSITY = 1.04 (air = 1) 1.58 1.53 Nitrogen oxides refer to several compounds composed of nitrogen and oxygen. Th is entry focuses on the three most common: nitric oxide, nitrogen dioxide, and nitrous oxide. Other nitrogen oxides not addressed include dinitrogen trioxide (N2O3), dinitrogen tetroxide (N2O4), nitrogen trioxide (NO3), and dinitrogen pentoxide (N2O5). As air pollutants, nitrogen oxides generally refer to the compounds nitric oxide and nitrogen dioxide. Nitrogen oxides exist as gaseous compounds under normal conditions, although nitrogen dioxide changes phases between liquid and gas near room temperature (its liquid density at 20°C is 1.45 g/cm3). Nitrogen monoxide (NO) is commonly called nitric oxide, and dinitrogen monoxide’s (N2O) common name is nitrous oxide. Nitric oxide and nitrogen dioxide are nonfl ammable, toxic gases. Nitric oxide is colorless and has a sharp sweet odor; nitrogen dioxide is a reddish-brown gas (or yellow liquid) with a strong, acrid odor. Nitrogen dioxide readily dimerizes to produce N2O4. Dinitrogen tetroxide production is favored as temperature decreases. -

SN2 Reactions of N2O5 with Ions in Water: Microscopic Mechanisms, Intermediates, and Products Natalia V
pubs.acs.org/JPCA Article SN2 Reactions of N2O5 with Ions in Water: Microscopic Mechanisms, Intermediates, and Products Natalia V. Karimova, James Chen, Joseph R. Gord, Sean Staudt, Timothy H. Bertram, Gilbert M. Nathanson, and R. Benny Gerber* Cite This: J. Phys. Chem. A 2020, 124, 711−720 Read Online ACCESS Metrics & More Article Recommendations ff ABSTRACT: Reactions of dinitrogen pentoxide (N2O5) greatly a ect the concentrations of NO3, ozone, OH radicals, methane, and more. In this work, we employ ab initio molecular dynamics and other tools of computational chemistry to explore reactions of N2O5 with anions hydrated by 12 water molecules to shed light on this important class of reactions. The ions − 2− − − investigated are Cl ,SO4 , ClO4 , and RCOO (R = H, CH3,C2H5). The following main results are obtained: (i) all the reactions take place by an SN2- type mechanism, with a transition state that involves a contact ion pair + − (NO2 NO3 ) that interacts strongly with water molecules. (ii) Reactions of a + solvent-separated nitronium ion (NO2 ) are not observed in any of the cases. (iii) An explanation is provided for the suppression of ClNO formation from N O reacting with salty water when sulfate or acetate ions are present, as found in recent experiments. (iv) 2 2 5 − Formation of novel intermediate species, such as (SO4NO2 ) and RCOONO2, in these reactions is predicted. The results suggest atomistic-level mechanisms for the reactions studied and may be useful for the development of improved modeling of reaction kinetics in aerosol particles. 1. INTRODUCTION theoretical study of hydrolysis of N2O5 molecules in small water clusters (five water molecules) was performed by Hillier Heterogeneous chemical reactions at atmospheric aqueous 24 interfaces between gas-phase molecules and the liquid-phase and co-workers. -
Binary Ionic Compounds : Metal and Non-Metal
Binary Ionic Compounds : Metal and Non-metal Formulas: Use oxidation numbers, Names: End in –ide, metals may have roman numerals. Examples: 1. Potassium Bromide KBr 2. Calcium Bromide CaBr2 3. Lithium Fluoride LiF 4. Lithium Oxide Li2O 5. Cu2O Copper (I) oxide 6. CuO Copper (II) oxide 7. FeCl2 Iron (II) chloride 8. FeCl3 Iron (III) chloride Try these: 1. MgO 2. BaS 3. K3P 4. Na3N 5. SnCl4 6. Mn2O3 7. PbS 8. Silver Fluoride 9. Calcium nitride 10. Sodium phosphide 11. Cobalt (II) bromide 12. Zinc Oxide 13. Aluminum sulfide 14. Manganese (II) chloride Answers: 1. Magnesium oxide 2. Barium sulfide 3. Potassium phosphide 4. Sodium nitride 5. Tin (IV) Chloride 6. Manganese (III) oxide 7. Lead (II) sulfide 8. AgF 9. Ca3N2 10. Na3P 11. CoBr2 12. ZnO 13. Al2S3 14. MnCl2 Binary Molecular Compounds: 2 non-metals Formulas: use prefixes to tell number of each kind of atom Names: formulas use prefixes to tell number of each kind of atom, end in --ide Examples: 1. CO carbon monoxide 2. CO2 carbon dioxide 3. SO2 sulfur dioxide 4. SO3 sulfur trioxide 5. N2H4 dinitrogen tetrahydride 6. silicon dioxide SiO2 7. phosphorus trichloride PCl3 8. Dinitrogen trioxide N2O3 9. Dinitrogen pentoxide N2O5 10. Carbon tetrachloride CCl4 Try these: 1. N2O5 2. PCl5 3. CS2 4. NO 5. N2O 6. SiF6 7. PO5 8. Carbon tetrabromide 9. Sulfur hexafluoride 10. Nitrogen dioxide 11. Dinitrogen heptaoxide 12. Boron monophosphide 13. Sulfur dioxide 14. Phosphorus trioxide Answers: 1. Dinitrogen pentoxide 2. Phosphorus pentachloride 3. Carbon disulfide 4. Nitrogen monoxide 5. Dinitrogen monoxide 6. -
Formulas and Names of Binary Covalent Compounds
FORMULAS AND NAMES OF BINARY COVALENT COMPOUNDS Fill the blanks in the following table. Formula Name Formula Name CO tetraphosphorus decaoxide carbon monoxide P4O10 carbon disulfide NH CS2 3 ammonia SO sulfur hexafluoride 2 sulfur dioxide SF6 SiC silicon carbide N2O3 dinitrogen trioxide dinitrogen monoxide OF N2O 2 oxygen difluoride silicon tetrachloride P4S3 tetraphosphorus trisulfide SiCl4 IF7 iodine heptafluoride CH4 methane sulfur trioxide H2O water SO3 dihydrogen disulfide H2S2 KrF2 krypton difluoride dinitrogen pentaoxide Cl2O7 dichlorine heptaoxide N2O5 FORMULAS AND NAMES OF BINARY IONIC COMPOUNDS (1) Write the formula for each ion below the ion’s name. (2) Fill in the blanks with the correct formula and name of the compound formed by the combination of ions. Chloride ion Oxide ion Bromide Sulfide ion Nitride ion ─ Cl 2─ ─ 2─ 3─ O Br S N Sodium ion NaBr NaCl Na O Na S Na N 2 2 3 + Na sodium chloride sodium oxide sodium bromide sodium sulfide sodium nitride Copper (I) ion CuCl Cu O CuBr Cu S Cu N 2 2 3 + Cu copper (I) chloride copper (I) oxide copper (I) bromide copper (I) sulfide copper (I) nitride Calcium ion CaCl CaO CaBr CaS Ca N 2 2 3 2 2+ calcium chloride calcium oxide calcium bromide calcium sulfide calcium nitride Ca Iron (II) ion FeCl FeO FeBr FeS Fe N 2 2 3 2 2+ iron (II) oxide iron (II) sulfide Fe iron (II) chloride iron (II) bromide iron (II) nitride Iron (III) ion FeCl Fe O FeBr Fe S FeN 3 2 3 3 2 3 3+ iron (III) nitride Fe iron (III) chloride iron (III) oxide iron (III) bromide iron (III) sulfide Aluminum ion AlCl Al O AlBr Al S AlN 3 2 3 3 2 3 3+ Al aluminum chloride aluminum oxide aluminum bromide aluminum sulfide aluminum nitride FORMULAS OF IONIC COMPOUNDS WITH MONO- AND POLYATOMIC IONS For each box write the chemical formula of the compound formed by the cation at the head of the column and the anion at the left of the row.