The Dynamics of E1A in Regulating Networks and Canonical Pathways
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Ailanthone Inhibits Non-Small Cell Lung Cancer Cell Growth Through Repressing DNA Replication Via Downregulating RPA1
FULL PAPER British Journal of Cancer (2017) 117, 1621–1630 | doi: 10.1038/bjc.2017.319 Keywords: ailanthone; non-small cell lung cancer; DNA replication; RPA1; Chinese medicine Ailanthone inhibits non-small cell lung cancer cell growth through repressing DNA replication via downregulating RPA1 Zhongya Ni1, Chao Yao1, Xiaowen Zhu1, Chenyuan Gong1, Zihang Xu2, Lixin Wang3, Suyun Li4, Chunpu Zou2 and Shiguo Zhu*,1,3 1Laboratory of Integrative Medicine, School of Basic Medical Sciences, Shanghai University of Traditional Chinese Medicine, 1200 Cai Lun Rd, Shanghai 201203, PR China; 2Department of Internal Classic of Medicine, School of Basic Medical Sciences, Shanghai University of Traditional Chinese Medicine, 1200 Cai Lun Rd, Shanghai 201203, PR China; 3Department of Immunology and Pathogenic Biology, School of Basic Medical Sciences, Shanghai University of Traditional Chinese Medicine, 1200 Cai Lun Rd, Shanghai 201203, PR China and 4Department of Pathology, School of Basic Medical Sciences, Shanghai University of Traditional Chinese Medicine, 1200 Cai Lun Rd, Shanghai 201203, PR China Background: The identification of bioactive compounds from Chinese medicine plays a crucial role in the development of novel reagents against non-small cell lung cancer (NSCLC). Methods: High throughput screening assay and analyses of cell growth, cell cycle, apoptosis, cDNA microarray, BrdU incorporation and gene expression were performed. Results: Ailanthone (Aila) suppressed NSCLC cell growth and colony formation in vitro and inhibited NSCLC tumour growth in subcutaneously xenografted and orthotopic lung tumour models, leading to prolonged survival of tumour-bearing mice. Moreover, Aila induced cell cycle arrest in a dose-independent manner but did not induce apoptosis in all NSCLC cells. -

Clipped Histone H3 Is Integrated Into Nucleosomes of DNA Replication
Clipped histone H3 is integrated into nucleosomes of DNA replication genes in the human malaria parasite Plasmodium falciparum Abril Marcela Herrera-solorio, Shruthi Sridhar Vembar, Cameron Ross Macpherson, Daniela Lozano-amado, Gabriela Romero Meza, Beatriz Xoconostle-cazares, Rafael Miyazawa Martins, Patty Chen, Miguel Vargas, Artur Scherf, et al. To cite this version: Abril Marcela Herrera-solorio, Shruthi Sridhar Vembar, Cameron Ross Macpherson, Daniela Lozano- amado, Gabriela Romero Meza, et al.. Clipped histone H3 is integrated into nucleosomes of DNA replication genes in the human malaria parasite Plasmodium falciparum. EMBO Reports, EMBO Press, 2019, 20 (4), pp.e46331. 10.15252/embr.201846331. hal-02321832 HAL Id: hal-02321832 https://hal.archives-ouvertes.fr/hal-02321832 Submitted on 9 Sep 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution| 4.0 International License Scientific Report Clipped histone H3 is integrated into nucleosomes of DNA replication genes in the human malaria parasite -

Anti-RPA1 Monoclonal Antibody, Clone 3I21 (DCABH-3680) This Product Is for Research Use Only and Is Not Intended for Diagnostic Use
Anti-RPA1 Monoclonal antibody, clone 3I21 (DCABH-3680) This product is for research use only and is not intended for diagnostic use. PRODUCT INFORMATION Product Overview Mouse monoclonal to RPA70 Antigen Description Plays an essential role in several cellular processes in DNA metabolism including replication, recombination and DNA repair. Binds and subsequently stabilizes single-stranded DNA intermediates and thus prevents complementary DNA from reannealing.Functions as component of the alternative replication protein A complex (aRPA). aRPA binds single-stranded DNA and probably plays a role in DNA repair; it does not support chromosomal DNA replication and cell cycle progression through S-phase. In vitro, aRPA cannot promote efficient priming by DNA polymerase alpha but supports DNA polymerase delta synthesis in the presence of PCNA and replication factor C (RFC), the dual incision/excision reaction of nucleotide excision repair and RAD51-dependent strand exchange. Immunogen Purified Human RPA70 protein Isotype IgG1 Source/Host Mouse Species Reactivity Human Clone 3I21 Purity Protein G purified Conjugate Unconjugated Applications WB, IP Positive Control HeLa cells Format Liquid Size 500 μl Buffer pH: 7.40; Preservative: 0.09% Sodium azide; Constituents: 99% PBS, 0.02% BSA Preservative 0.09% Sodium Azide Storage Store at +4°C short term (1-2 weeks). Store at -20°C or -80°C. Avoid freeze / thaw cycle. 45-1 Ramsey Road, Shirley, NY 11967, USA Email: [email protected] Tel: 1-631-624-4882 Fax: 1-631-938-8221 1 © Creative Diagnostics -

CHK1 Inhibition Is Synthetically Lethal with Loss of B-Family DNA Polymerase Function in Human Lung and Colorectal Cancer Cells
Author Manuscript Published OnlineFirst on March 11, 2020; DOI: 10.1158/0008-5472.CAN-19-1372 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. 1 Title: CHK1 inhibition is synthetically lethal with loss of B-family DNA polymerase function in human lung and colorectal cancer cells. Author List: Rebecca F. Rogers1, Mike I. Walton1, Daniel L. Cherry2, Ian Collins1, Paul A. Clarke1, Michelle D. Garrett2* and Paul Workman1* Affiliations: 1Cancer Research UK Cancer Therapeutics Unit, The Institute of Cancer Research, London, SM2 5NG, UK 2 School of Biosciences, Stacey Building, University of Kent, Canterbury, Kent, CT2 7NJ, UK Running title: Synthetic lethality of CHK1 and DNA polymerase inhibition *Corresponding authors: Michelle D Garrett and Paul Workman Corresponding Author Information Michelle D Garrett School of Biosciences, Stacey Building, University of Kent, Canterbury, Kent, CT2 7NJ, UK. Email: [email protected] Phone: +44 (0)1227 816140 Paul Workman Cancer Research UK Cancer Therapeutics Unit, The Institute of Cancer Research, London, SM2 5NG, UK. Email: [email protected] Phone: +44 (0)20 7153 5209 Conflict of interest statement: IC, MDG, MIW, RFR, PC and PW are current or past employees of The Institute of Cancer Research, which has a commercial interest in the discovery and development of CHK1 inhibitors, including SRA737, and operates a rewards-to- inventors scheme. IC, MDG and MIW have been involved in a commercial collaboration on CHK1 inhibitors with Sareum Ltd and intellectual property arising from the program, including SRA737, was licensed to Sierra Oncology. IC is a consultant for Sierra Oncology, Adorx Ltd, Epidarex LLP and Enterprise Therapeutics Ltd and holds equity in Monte Rosa Therapeutics AG. -

Expression of an Ortholog of Replication Protein A1 (RPA1)
Proc. Natl. Acad. Sci. USA Vol. 94, pp. 9979–9983, September 1997 Plant Biology Expression of an ortholog of replication protein A1 (RPA1) is induced by gibberellin in deepwater rice (cell divisionyintercalary meristemyinternodal growthyOryza sativa) ESTHER VAN DER KNAAP*, SANDRINE JAGOUEIX*†, AND HANS KENDE‡ Michigan State University–Department of Energy Plant Research Laboratory, Michigan State University, East Lansing, MI 48824-1312 Contributed by Hans Kende, July 16, 1997 ABSTRACT Internodes of deepwater rice are induced to antagonist of GA action in rice (6). Growth of the internode grow rapidly when plants become submerged. This adaptation is, ultimately, promoted by GA (5). enables deepwater rice to keep part of its foliage above the The primary target tissue of GA is the intercalary meristem rising flood waters during the monsoon season and to avoid of the internode, where GA enhances cell division activity and drowning. This growth response is, ultimately, elicited by the cell elongation (5, 7, 8). The intercalary meristem is a zone of plant hormone gibberellin (GA). The primary target tissue for about 3 mm in length and is located at the base of the internode GA action is the intercalary meristem of the internode. Using (9). Cells are displaced from the intercalary meristem into the differential display of mRNA, we have isolated a number of elongation zone, where they reach their final size. In GA- genes whose expression in the intercalary meristem is regu- treated internodes, the final cell length is about three to four lated by GA. The product of one of these genes was identified times longer than in control internodes (5, 7). -

Identification of Proteins Involved in the Maintenance of Genome Stability
Identification of Proteins Involved in the Maintenance of Genome Stability by Edith Hang Yu Cheng A thesis submitted in conformity with the requirements for the degree of Doctor of Philosophy Department of Biochemistry University of Toronto ©Copyright by Edith Cheng2015 Identification of Proteins Involved in the Maintenance of Genome Stability Edith Cheng Doctor of Philosophy Department of Biochemistry University of Toronto 2015 Abstract Aberrant changes to the genome structure underlie numerous human diseases such as cancers. The functional characterization ofgenesand proteins that maintain chromosome stability will be important in understanding disease etiology and developing therapeutics. I took a multi-faceted approach to identify and characterize genes involved in the maintenance of genome stability. As biological pathways involved in genome maintenance are highly conserved in evolution, results from model organisms can greatly facilitate functional discovery in humans. In S. cerevisiae, I identified 47 essential gene depletions with elevated levels of spontaneous DNA damage foci and 92 depletions that caused elevated levels of chromosome rearrangements. Of these, a core subset of 15 DNA replication genes demonstrated both phenotypes when depleted. Analysis of rearrangement breakpoints revealed enrichment at yeast fragile sites, Ty retrotransposons, early origins of replication and replication termination sites. Together, thishighlighted the integral role of DNA replicationin genome maintenance. In light of my findings in S. cerevisiae, I identified a list of 153 human proteins that interact with the nascentDNA at replication forks, using a DNA pull down strategy (iPOND) in human cell lines. As a complementary approach for identifying human proteins involved in genome ii maintenance, I usedthe BioID techniqueto discernin vivo proteins proximal to the human BLM- TOP3A-RMI1-RMI2 genome stability complex, which has an emerging role in DNA replication progression. -

A Conserved but Plant-Specific CDK-Mediated Regulation of DNA Replication Protein A2 in the Precise Control of Stomatal Terminal Division
A conserved but plant-specific CDK-mediated regulation of DNA replication protein A2 in the precise control of stomatal terminal division Kezhen Yanga, Lingling Zhua,b, Hongzhe Wanga, Min Jianga, Chunwang Xiaoc,d, Xiangyang Hue, Steffen Vannestef,g,h, Juan Dongi, and Jie Lea,b,1 aKey Laboratory of Plant Molecular Physiology, CAS Center for Excellence in Molecular Plant Sciences, Institute of Botany, Chinese Academy of Sciences, 100093 Beijing, China; bUniversity of Chinese Academy of Sciences, 100049 Beijing, China; cCollege of Life and Environmental Sciences, Minzu University of China, 100081 Beijing, China; dHulun Lake Reserve Grassland Ecology Research Station, Minzu University of China, 100081 Beijing, China; eShanghai Key Laboratory of Bio-Energy Crops, School of Life Sciences, Shanghai University, 200444 Shanghai, China; fCenter for Plant Systems Biology, VIB, 9052 Ghent, Belgium; gDepartment of Plant Biotechnology and Bioinformatics, Ghent University, 9052 Ghent, Belgium; hLaboratory of Plant Growth Analysis, Ghent University Global Campus, 21985 Incheon, Republic of Korea; and iWaksman Institute of Microbiology, Rutgers, The State University of New Jersey, Piscataway, NJ 08854 Edited by David C. Baulcombe, University of Cambridge, Cambridge, United Kingdom, and approved July 29, 2019 (received for review November 11, 2018) The R2R3-MYB transcription factor FOUR LIPS (FLP) controls the condition, RPA2 is hyperphosphorylated by the PIKK-family stomatal terminal division through transcriptional repression of kinases (ATM, ATR, and DNA-PK) that facilitates mitotic exit the cell cycle genes CYCLIN-DEPENDENT KINASE (CDK) B1s (CDKB1s), and the initiation of DNA repair (13–15). All known RPA2 CDKA;1,andCYCLIN A2s (CYCA2s). We mutagenized the weak mu- homologs have a conserved N-terminal phosphorylation domain, tant allele flp-1 seeds with ethylmethane sulfonate and screened although the specific residues may be not conserved in different out a flp-1 suppressor 1 (fsp1) that suppressed the flp-1 stomatal species (11). -
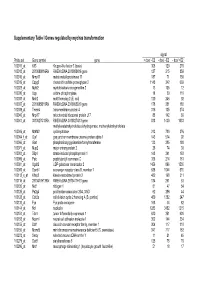
Supplementary Table I Genes Regulated by Myc/Ras Transformation
Supplementary Table I Genes regulated by myc/ras transformation signal Probe set Gene symbol gene + dox/ - E2 - dox/ - E2 - dox/ +E2 100011_at Klf3 Kruppel-like factor 3 (basic) 308 120 270 100013_at 2010008K16Rik RIKEN cDNA 2010008K16 gene 127 215 859 100016_at Mmp11 matrix metalloproteinase 11 187 71 150 100019_at Cspg2 chondroitin sulfate proteoglycan 2 1148 342 669 100023_at Mybl2 myeloblastosis oncogene-like 2 13 105 12 100030_at Upp uridine phosphorylase 18 39 110 100033_at Msh2 mutS homolog 2 (E. coli) 120 246 92 100037_at 2310005B10Rik RIKEN cDNA 2310005B10 gene 178 351 188 100039_at Tmem4 transmembrane protein 4 316 135 274 100040_at Mrpl17 mitochondrial ribosomal protein L17 88 142 89 100041_at 3010027G13Rik RIKEN cDNA 3010027G13 gene 818 1430 1033 methylenetetrahydrofolate dehydrogenase, methenyltetrahydrofolate 100046_at Mthfd2 cyclohydrolase 213 720 376 100064_f_at Gja1 gap junction membrane channel protein alpha 1 143 574 92 100066_at Gart phosphoribosylglycinamide formyltransferase 133 385 180 100071_at Mup2 major urinary protein 2 26 74 30 100081_at Stip1 stress-induced phosphoprotein 1 148 341 163 100089_at Ppic peptidylprolyl isomerase C 358 214 181 100091_at Ugalt2 UDP-galactose translocator 2 1456 896 1530 100095_at Scarb1 scavenger receptor class B, member 1 638 1044 570 100113_s_at Kifap3 kinesin-associated protein 3 482 168 311 100116_at 2810417H13Rik RIKEN cDNA 2810417H13 gene 134 261 53 100120_at Nid1 nidogen 1 81 47 54 100125_at Pa2g4 proliferation-associated 2G4, 38kD 62 286 44 100128_at Cdc2a cell division cycle 2 homolog A (S. pombe) 459 1382 247 100133_at Fyn Fyn proto-oncogene 168 44 82 100144_at Ncl nucleolin 1283 3452 1215 100151_at Tde1 tumor differentially expressed 1 620 351 620 100153_at Ncam1 neural cell adhesion molecule 1 302 144 234 100155_at Ddr1 discoidin domain receptor family, member 1 304 117 310 100156_at Mcmd5 mini chromosome maintenance deficient 5 (S. -

DNA Repair Deficiency in Huntington's Disease Fibroblasts and Induced Pluripotent Stem Cells
Old Dominion University ODU Digital Commons Biological Sciences Theses & Dissertations Biological Sciences Fall 2015 DNA Repair Deficiency in Huntington's Disease Fibroblasts and Induced Pluripotent Stem Cells Peter Anthony Mollica Old Dominion University, [email protected] Follow this and additional works at: https://digitalcommons.odu.edu/biology_etds Part of the Cell Biology Commons, Developmental Biology Commons, and the Molecular Biology Commons Recommended Citation Mollica, Peter A.. "DNA Repair Deficiency in Huntington's Disease Fibroblasts and Induced Pluripotent Stem Cells" (2015). Doctor of Philosophy (PhD), Dissertation, Biological Sciences, Old Dominion University, DOI: 10.25777/r105-c396 https://digitalcommons.odu.edu/biology_etds/7 This Dissertation is brought to you for free and open access by the Biological Sciences at ODU Digital Commons. It has been accepted for inclusion in Biological Sciences Theses & Dissertations by an authorized administrator of ODU Digital Commons. For more information, please contact [email protected]. DNA REPAIR DEFICIENCY IN HUNTINGTON’S DISEASE FIBROBLASTS AND INDUCED PLURIPOTENT STEM CELLS by Peter A. Mollica B.S. December 2011, Old Dominion University A Dissertation Submitted to the Faculty of Old Dominion University in Partial Fulfillment of the Requirements for the Degree of DOCTOR OF PHILOSOPHY BIOMEDICAL SCIENCES OLD DOMINION UNIVERSITY December 2015 Approved by : ______________________________ Christopher J. Osgood (Co-Advisor) ______________________________ Roy C. Ogle (Co-Advisor) ______________________________ Lesley H. Greene (Member) ABSTRACT DNA REPAIR DEFICIENCY IN HUNTINGTON’S DISEASE FIBROBLASTS AND INDUCED PLURIPOTENT STEM CELLS Peter A. Mollica Old Dominion University, 2015 Co-Advisors: Dr. Christopher Osgood Dr. Roy C. Ogle Mutant huntingtin protein (mhtt)–the protein responsible for cellular dysfunction in Huntington’s disease (HD) –is a product of an expanded trinucleotide repeat (TNR) cytosine-adenine-guanine (CAG) sequence in exon 1 of the huntingtin (HTT) gene. -

CHK1 Inhibition Is Synthetically Lethal with Loss of B-Family DNA Polymerase Function in Human Lung and Colorectal Cancer Cells a C Rebecca F
Published OnlineFirst March 11, 2020; DOI: 10.1158/0008-5472.CAN-19-1372 CANCER RESEARCH | TRANSLATIONAL SCIENCE CHK1 Inhibition Is Synthetically Lethal with Loss of B-Family DNA Polymerase Function in Human Lung and Colorectal Cancer Cells A C Rebecca F. Rogers1, Michael I. Walton1, Daniel L. Cherry2, Ian Collins1, Paul A. Clarke1, Michelle D. Garrett2, and Paul Workman1 ABSTRACT ◥ Checkpoint kinase 1 (CHK1) is a key mediator of the DNA Replication stress, DNA damage, and apoptosis were increased in damage response that regulates cell-cycle progression, DNA human cancer cells following depletion of the B-family DNA damage repair, and DNA replication. Small-molecule CHK1 polymerases combined with SRA737 treatment. Moreover, phar- inhibitors sensitize cancer cells to genotoxic agents and have macologic blockade of B-family DNA polymerases using aphi- shown single-agent preclinical activity in cancers with high levels dicolin or CD437 combined with CHK1 inhibitors led to syn- of replication stress. However, the underlying genetic determi- ergistic inhibition of cancer cell proliferation. Furthermore, low nants of CHK1 inhibitor sensitivity remain unclear. We used the levels of POLA1, POLE, and POLE2 protein expression in developmental clinical drug SRA737 in an unbiased large-scale NSCLC and colorectal cancer cells correlated with single-agent siRNA screen to identify novel mediators of CHK1 inhibitor CHK1 inhibitor sensitivity and may constitute biomarkers of this sensitivity and uncover potential combination therapies and phenotype. These findings provide a potential basis for combin- biomarkers for patient selection. We identified subunits of the ing CHK1 and B-family polymerase inhibitors in cancer therapy. B-family of DNA polymerases (POLA1, POLE,andPOLE2) whose silencing sensitized the human A549 non–small cell lung Significance: These findings demonstrate how the therapeutic cancer (NSCLC) and SW620 colorectal cancer cell lines to benefit of CHK1 inhibitors may potentially be enhanced and could SRA737. -

HPV Microarrays
Supplementary Methods Tissue sample process. Each tissue sample was cryosectioned, and selected sections stained with hematoxylin/eosin, and reviewed to determine tumor content, pathological status, and freedom from necrosis and freezing artifacts. Epithelial cells from all normal samples and tumor cells from HNC or CC samples with less than 80% tumor were laser capture microdissected from adjacent sections using a PixCell II LCM system (Arcturus, Mountain View, CA) (Supplementary Table 2). For guidance, an adjacent section was briefly stained with hematoxylin to visualize tissue structure. RNA amplification and labeling. Total RNA was extracted from sectioned and/or microdissected samples as follows: 1 ml of TRIzol (Invitrogen, Carlsbad, CA) was added to each tissue sample, homogenized by passing through a 20 gauge needle, and added to 0.2 ml chloroform. After centrifugation at 20,000 xg for 20 min at 4oC, RNA in the aqueous phase was precipitated with an equal volume of isopropanol for 30 min at 4oC, pelleted, and washed twice with cold 70% ethanol. Double strand (ds) cDNA was synthesized from this RNA using a SuperScript ds cDNA synthesis kit (Invitrogen) and T7 promoter-linked oligo (dT)24. Complementary RNA (cRNA) was synthesized from T7 promoter-linked ds cDNA using a MEGAscript high transcription kit (Ambion, Austin, TX). To obtain a sufficient cRNA for ≥2 microarray hybridizations, this amplification process was repeated. Second round cRNA was biotin labeled using a BioArray High Yield RNA Transcript Labeling Kit (Enzo Life Sciences, Farmingdale, NY) and stored at -80oC until hybridized. cRNA quality and quantity was determined by gel electrophoresis and UV spectrophotometry. -

(12) Patent Application Publication (10) Pub. No.: US 2011/0196614 A1 Banchereau Et Al
US 2011 0196614A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2011/0196614 A1 Banchereau et al. (43) Pub. Date: Aug. 11, 2011 (54) BLOODTRANSCRIPTIONAL SIGNATURE OF Related U.S. Application Data MYCOBACTERIUMITUBERCULOSS INFECTION (60) Provisional application No. 61/075,728, filed on Jun. 25, 2008. (75) Inventors: Jacques F. Banchereau, Dallas, TX Publication Classification (US); Damien Chaussabel, Bainbridge Island, WA (US); Anne (51) Int. Cl. O'Garra, London (GB); Matthew G06F 9/00 (2011.01) Berry, London (GB); Onn Min GOIN 33/48 (2006.01) Kon, London (GB) (52) U.S. Cl. .......................................................... 702/19 (57) ABSTRACT (73) Assignees: BAYLOR RESEARCH INSTITUTE, Dallas, TX (US); The present invention includes methods, systems and kits for NATIONAL INSTITUTE FOR distinguishing between active and latent mycobacterium MEDICAL RESEARCH, London tuberculosis infection in a patient Suspected of being infected (GB); IMPERIAL COLLEGE with mycobacterium tuberculosis, and distinguishing Such HEALTHCARE NHS TRUST, patients from uninfected individuals, the method including London (GB) the steps of obtaining a gene expression dataset from a whole blood obtained sample from the patient and determining the (21) Appl. No.: 12/602,488 differential expression of one or more transcriptional gene expression modules that distinguish between infected and (22) PCT Fled: Jun. 25, 2009 non-infected patients, wherein the dataset demonstrates an aggregate change in the levels of polynucleotides in the one or (86) PCT NO.: PCT/USO9A48698 more transcriptional gene expression modules as compared to matched non-infected patients, thereby distinguishing S371 (c)(1), between active and latent mycobacterium tuberculosis infec (2), (4) Date: Apr. 25, 2011 tion.