Identification of Proteins Involved in the Maintenance of Genome Stability
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Functional Compensation Among HMGN Variants Modulates the Dnase I Hypersensitive Sites at Enhancers
Downloaded from genome.cshlp.org on October 9, 2021 - Published by Cold Spring Harbor Laboratory Press Research Functional compensation among HMGN variants modulates the DNase I hypersensitive sites at enhancers Tao Deng,1,12 Z. Iris Zhu,2,12 Shaofei Zhang,1 Yuri Postnikov,1 Di Huang,2 Marion Horsch,3 Takashi Furusawa,1 Johannes Beckers,3,4,5 Jan Rozman,3,5 Martin Klingenspor,6,7 Oana Amarie,3,8 Jochen Graw,3,8 Birgit Rathkolb,3,5,9 Eckhard Wolf,9 Thure Adler,3 Dirk H. Busch,10 Valérie Gailus-Durner,3 Helmut Fuchs,3 Martin Hrabeˇ de Angelis,3,4,5 Arjan van der Velde,2,13 Lino Tessarollo,11 Ivan Ovcherenko,2 David Landsman,2 and Michael Bustin1 1Protein Section, Laboratory of Metabolism, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, Maryland 20892, USA; 2Computational Biology Branch, National Center for Biotechnology Information, National Library of Medicine, Bethesda, Maryland 20892, USA; 3German Mouse Clinic, Institute of Experimental Genetics, Helmholtz Zentrum München, German Research Center for Environmental Health, 85764 Neuherberg, Germany; 4Experimental Genetics, Center of Life and Food Sciences Weihenstephan, Technische Universität München, 85354 Freising-Weihenstephan, Germany; 5German Center for Diabetes Research (DZD), 85764 Neuherberg, Germany; 6Molecular Nutritional Medicine, Technische Universität München, 85350 Freising, Germany; 7Center for Nutrition and Food Sciences, Technische Universität München, 85350 Freising, Germany; 8Institute of Developmental Genetics (IDG), 85764 -

Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model
Downloaded from http://www.jimmunol.org/ by guest on September 25, 2021 T + is online at: average * The Journal of Immunology , 34 of which you can access for free at: 2016; 197:1477-1488; Prepublished online 1 July from submission to initial decision 4 weeks from acceptance to publication 2016; doi: 10.4049/jimmunol.1600589 http://www.jimmunol.org/content/197/4/1477 Molecular Profile of Tumor-Specific CD8 Cell Hypofunction in a Transplantable Murine Cancer Model Katherine A. Waugh, Sonia M. Leach, Brandon L. Moore, Tullia C. Bruno, Jonathan D. Buhrman and Jill E. Slansky J Immunol cites 95 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2016/07/01/jimmunol.160058 9.DCSupplemental This article http://www.jimmunol.org/content/197/4/1477.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2016 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 25, 2021. The Journal of Immunology Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model Katherine A. -

How Shelterin Protects Mammalian Telomeres 303 ANRV361-GE42-15 ARI 3 October 2008 10:10 (See 3' 5' TRF2 S Mplex
ANRV361-GE42-15 ARI 3 October 2008 10:10 ANNUAL How Shelterin Protects REVIEWS Further Click here for quick links to Annual Reviews content online, Mammalian Telomeres including: • Other articles in this volume 1 • Top cited articles Wilhelm Palm and Titia de Lange • Top downloaded articles • Our comprehensive search Laboratory for Cell Biology and Genetics, The Rockefeller University, New York, NY 10021; email: [email protected] 1Current address: Max Planck Institute of Molecular Cell Biology and Genetics, 01307 Dresden, Germany Annu. Rev. Genet. 2008. 42:301–34 Key Words First published online as a Review in Advance on ATM, ATR, cancer, NHEJ, HR August 4, 2008 by Rockefeller University on 10/13/09. For personal use only. The Annual Review of Genetics is online at Abstract genet.annualreviews.org The genomes of prokaryotes and eukaryotic organelles are usually cir- This article’s doi: cular as are most plasmids and viral genomes. In contrast, the nuclear Annu. Rev. Genet. 2008.42:301-334. Downloaded from arjournals.annualreviews.org 10.1146/annurev.genet.41.110306.130350 genomes of eukaryotes are organized on linear chromosomes, which Copyright c 2008 by Annual Reviews. require mechanisms to protect and replicate DNA ends. Eukaryotes All rights reserved navigate these problemswith the advent of telomeres, protective nucle- 0066-4197/08/1201-0301$20.00 oprotein complexes at the ends of linear chromosomes, and telomerase, the enzyme that maintains the DNA in these structures. Mammalian telomeres contain a specific protein complex, shelterin, that functions to protect chromosome ends from all aspects of the DNA damage re- sponse and regulates telomere maintenance by telomerase. -

Pan-Cancer Analysis of Alternative Lengthening of Telomere Activity
cancers Article Pan-Cancer Analysis of Alternative Lengthening of Telomere Activity Ji-Yong Sung 1,2, Hee-Woong Lim 3, Je-Gun Joung 1,* and Woong-Yang Park 1,2,4,* 1 Samsung Genome Institute, Samsung Medical Center, Seoul 06351, Korea; [email protected] 2 Department of Health Science and Technology, Samsung Advanced Institute of Health Science and Technology, Sungkyunkwan University, Seoul 06351, Korea 3 Division of Biomedical Informatics, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH 45229, USA; [email protected] 4 Department of Molecular Cell Biology, School of Medicine, Sungkyunkwan University, Seoul 06351, Korea * Correspondence: [email protected] (J.-G.J.); [email protected] (W.-Y.P.); Tel.: +82-2-3410-1706 (J.-G.J.); +82-2-3410-6128 (W.-Y.P.); Fax: +82-2-2148-9819 (W.-Y.P.) Received: 30 June 2020; Accepted: 5 August 2020; Published: 7 August 2020 Abstract: Alternative lengthening of telomeres (ALT) is a telomerase-independent mechanism that extends telomeres in cancer cells. It influences tumorigenesis and patient survival. Despite the clinical significance of ALT in tumors, the manner in which ALT is activated and influences prognostic outcomes in distinct cancer types is unclear. In this work, we profiled distinct telomere maintenance mechanisms (TMMs) using 8953 transcriptomes of 31 different cancer types from The Cancer Genome Atlas (TCGA). Our results demonstrated that approximately 29% of cancer types display high ALT activity with low telomerase activity in the telomere-lengthening group. Among the distinct ALT mechanisms, homologous recombination was frequently observed in sarcoma, adrenocortical carcinoma, and kidney chromophobe. Five cancer types showed a significant difference in survival in the presence of high ALT activity. -

Iron Depletion Reduces Abce1 Transcripts While Inducing The
Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 22 October 2019 doi:10.20944/preprints201910.0252.v1 1 Research Article 2 Iron depletion Reduces Abce1 Transcripts While 3 Inducing the Mitophagy Factors Pink1 and Parkin 4 Jana Key 1,2, Nesli Ece Sen 1, Aleksandar Arsovic 1, Stella Krämer 1, Robert Hülse 1, Suzana 5 Gispert-Sanchez 1 and Georg Auburger 1,* 6 1 Experimental Neurology, Goethe University Medical School, 60590 Frankfurt am Main; 7 2 Faculty of Biosciences, Goethe-University Frankfurt am Main, Germany 8 * Correspondence: [email protected] 9 10 Abstract: Lifespan extension was recently achieved in Caenorhabditis elegans nematodes by 11 mitochondrial stress and mitophagy, triggered via iron depletion. Conversely in man, deficient 12 mitophagy due to Pink1/Parkin mutations triggers iron accumulation in patient brain and limits 13 survival. We now aimed to identify murine fibroblast factors, which adapt their mRNA expression 14 to acute iron manipulation, relate to mitochondrial dysfunction and may influence survival. After 15 iron depletion, expression of the plasma membrane receptor Tfrc with its activator Ireb2, the 16 mitochondrial membrane transporter Abcb10, the heme-release factor Pgrmc1, the heme- 17 degradation enzyme Hmox1, the heme-binding cholesterol metabolizer Cyp46a1, as well as the 18 mitophagy regulators Pink1 and Parkin showed a negative correlation to iron levels. After iron 19 overload, these factors did not change expression. Conversely, a positive correlation of mRNA levels 20 with both conditions of iron availability was observed for the endosomal factors Slc11a2 and Steap2, 21 as well as for the iron-sulfur-cluster (ISC)-containing factors Ppat, Bdh2 and Nthl1. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -
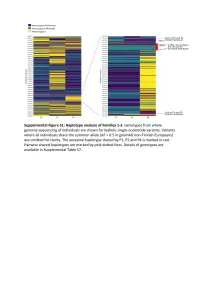
Supplementary Text and Figures.Pdf
Supplemental Figure S2. PRIM1 immunoblots (20-75 kDa). (A) Corresponds to Figure 2D, with additional bands seen in P2 at 75 kDa and 25 kDa. (B) Independent experiment with cell lysates from C and P2 in which additional bands at 25 kDa not evident. (C) Corresponds to Figure 2D. No additional band visible at 25 kDa. (D) Validation of monoclonal antibody (8G10) with siRNA to PRIM1 in HeLa cells. siLUC, luciferase negative control. siPRIM1, siRNA targeting PRIM1 transcript. Vinculin used as a loading control. Supplemental Figure S3: Predicted destabilisation of PRIM1 by the C301R substitution. (A) Cysteine 301 lies in a buried hydrophobic region in PRIM1. DNA primase dimer crystal structure (PDB: 4BPU). PRIM1 residues shaded according to solvent accessibility. Cysteine 301, substituted to arginine in P5, lies in a buried hydrophobic region. (B) Cys301 is evolutionarily conserved in vertebrates. Neither arginine or other large or charged amino acids are observed at this position in other species. (C) The C301R substitution observed in P5 is predicted to lead to destabilisation of all available PRIM1 crystal structures. In contrast, substitution with leucine or threonine, as observed in some orthologous proteins, is not predicted to lead to destabilisation. Data points, predicted changes in free-energy (ΔΔG) from FoldX plotted for each of 14 available crystal structures. (D) Immunoblotting of RPE1 cells transfected with dual reporter constructs (as described in Figure 3B and Materials and Methods) confirms production of PRIM1-GFP and FLAG-SR at expected molecular weights and shows reduced protein levels for the C301R variant. n=2 experiments shown. C301R protein levels relative to WT after normalization to actin loading control indicated underneath the blot. -

1 AGING Supplementary Table 2
SUPPLEMENTARY TABLES Supplementary Table 1. Details of the eight domain chains of KIAA0101. Serial IDENTITY MAX IN COMP- INTERFACE ID POSITION RESOLUTION EXPERIMENT TYPE number START STOP SCORE IDENTITY LEX WITH CAVITY A 4D2G_D 52 - 69 52 69 100 100 2.65 Å PCNA X-RAY DIFFRACTION √ B 4D2G_E 52 - 69 52 69 100 100 2.65 Å PCNA X-RAY DIFFRACTION √ C 6EHT_D 52 - 71 52 71 100 100 3.2Å PCNA X-RAY DIFFRACTION √ D 6EHT_E 52 - 71 52 71 100 100 3.2Å PCNA X-RAY DIFFRACTION √ E 6GWS_D 41-72 41 72 100 100 3.2Å PCNA X-RAY DIFFRACTION √ F 6GWS_E 41-72 41 72 100 100 2.9Å PCNA X-RAY DIFFRACTION √ G 6GWS_F 41-72 41 72 100 100 2.9Å PCNA X-RAY DIFFRACTION √ H 6IIW_B 2-11 2 11 100 100 1.699Å UHRF1 X-RAY DIFFRACTION √ www.aging-us.com 1 AGING Supplementary Table 2. Significantly enriched gene ontology (GO) annotations (cellular components) of KIAA0101 in lung adenocarcinoma (LinkedOmics). Leading Description FDR Leading Edge Gene EdgeNum RAD51, SPC25, CCNB1, BIRC5, NCAPG, ZWINT, MAD2L1, SKA3, NUF2, BUB1B, CENPA, SKA1, AURKB, NEK2, CENPW, HJURP, NDC80, CDCA5, NCAPH, BUB1, ZWILCH, CENPK, KIF2C, AURKA, CENPN, TOP2A, CENPM, PLK1, ERCC6L, CDT1, CHEK1, SPAG5, CENPH, condensed 66 0 SPC24, NUP37, BLM, CENPE, BUB3, CDK2, FANCD2, CENPO, CENPF, BRCA1, DSN1, chromosome MKI67, NCAPG2, H2AFX, HMGB2, SUV39H1, CBX3, TUBG1, KNTC1, PPP1CC, SMC2, BANF1, NCAPD2, SKA2, NUP107, BRCA2, NUP85, ITGB3BP, SYCE2, TOPBP1, DMC1, SMC4, INCENP. RAD51, OIP5, CDK1, SPC25, CCNB1, BIRC5, NCAPG, ZWINT, MAD2L1, SKA3, NUF2, BUB1B, CENPA, SKA1, AURKB, NEK2, ESCO2, CENPW, HJURP, TTK, NDC80, CDCA5, BUB1, ZWILCH, CENPK, KIF2C, AURKA, DSCC1, CENPN, CDCA8, CENPM, PLK1, MCM6, ERCC6L, CDT1, HELLS, CHEK1, SPAG5, CENPH, PCNA, SPC24, CENPI, NUP37, FEN1, chromosomal 94 0 CENPL, BLM, KIF18A, CENPE, MCM4, BUB3, SUV39H2, MCM2, CDK2, PIF1, DNA2, region CENPO, CENPF, CHEK2, DSN1, H2AFX, MCM7, SUV39H1, MTBP, CBX3, RECQL4, KNTC1, PPP1CC, CENPP, CENPQ, PTGES3, NCAPD2, DYNLL1, SKA2, HAT1, NUP107, MCM5, MCM3, MSH2, BRCA2, NUP85, SSB, ITGB3BP, DMC1, INCENP, THOC3, XPO1, APEX1, XRCC5, KIF22, DCLRE1A, SEH1L, XRCC3, NSMCE2, RAD21. -

HMGB1 in Health and Disease R
Donald and Barbara Zucker School of Medicine Journal Articles Academic Works 2014 HMGB1 in health and disease R. Kang R. C. Chen Q. H. Zhang W. Hou S. Wu See next page for additional authors Follow this and additional works at: https://academicworks.medicine.hofstra.edu/articles Part of the Emergency Medicine Commons Recommended Citation Kang R, Chen R, Zhang Q, Hou W, Wu S, Fan X, Yan Z, Sun X, Wang H, Tang D, . HMGB1 in health and disease. 2014 Jan 01; 40():Article 533 [ p.]. Available from: https://academicworks.medicine.hofstra.edu/articles/533. Free full text article. This Article is brought to you for free and open access by Donald and Barbara Zucker School of Medicine Academic Works. It has been accepted for inclusion in Journal Articles by an authorized administrator of Donald and Barbara Zucker School of Medicine Academic Works. Authors R. Kang, R. C. Chen, Q. H. Zhang, W. Hou, S. Wu, X. G. Fan, Z. W. Yan, X. F. Sun, H. C. Wang, D. L. Tang, and +8 additional authors This article is available at Donald and Barbara Zucker School of Medicine Academic Works: https://academicworks.medicine.hofstra.edu/articles/533 NIH Public Access Author Manuscript Mol Aspects Med. Author manuscript; available in PMC 2015 December 01. NIH-PA Author ManuscriptPublished NIH-PA Author Manuscript in final edited NIH-PA Author Manuscript form as: Mol Aspects Med. 2014 December ; 0: 1–116. doi:10.1016/j.mam.2014.05.001. HMGB1 in Health and Disease Rui Kang1,*, Ruochan Chen1, Qiuhong Zhang1, Wen Hou1, Sha Wu1, Lizhi Cao2, Jin Huang3, Yan Yu2, Xue-gong Fan4, Zhengwen Yan1,5, Xiaofang Sun6, Haichao Wang7, Qingde Wang1, Allan Tsung1, Timothy R. -

Bioinformatics-Based Screening of Key Genes for Transformation of Liver
Jiang et al. J Transl Med (2020) 18:40 https://doi.org/10.1186/s12967-020-02229-8 Journal of Translational Medicine RESEARCH Open Access Bioinformatics-based screening of key genes for transformation of liver cirrhosis to hepatocellular carcinoma Chen Hao Jiang1,2, Xin Yuan1,2, Jiang Fen Li1,2, Yu Fang Xie1,2, An Zhi Zhang1,2, Xue Li Wang1,2, Lan Yang1,2, Chun Xia Liu1,2, Wei Hua Liang1,2, Li Juan Pang1,2, Hong Zou1,2, Xiao Bin Cui1,2, Xi Hua Shen1,2, Yan Qi1,2, Jin Fang Jiang1,2, Wen Yi Gu4, Feng Li1,2,3 and Jian Ming Hu1,2* Abstract Background: Hepatocellular carcinoma (HCC) is the most common type of liver tumour, and is closely related to liver cirrhosis. Previous studies have focussed on the pathogenesis of liver cirrhosis developing into HCC, but the molecular mechanism remains unclear. The aims of the present study were to identify key genes related to the transformation of cirrhosis into HCC, and explore the associated molecular mechanisms. Methods: GSE89377, GSE17548, GSE63898 and GSE54236 mRNA microarray datasets from Gene Expression Omni- bus (GEO) were analysed to obtain diferentially expressed genes (DEGs) between HCC and liver cirrhosis tissues, and network analysis of protein–protein interactions (PPIs) was carried out. String and Cytoscape were used to analyse modules and identify hub genes, Kaplan–Meier Plotter and Oncomine databases were used to explore relationships between hub genes and disease occurrence, development and prognosis of HCC, and the molecular mechanism of the main hub gene was probed using Kyoto Encyclopedia of Genes and Genomes(KEGG) pathway analysis. -

Gene Expression Profiling Analysis Contributes to Understanding the Association Between Non-Syndromic Cleft Lip and Palate, and Cancer
2110 MOLECULAR MEDICINE REPORTS 13: 2110-2116, 2016 Gene expression profiling analysis contributes to understanding the association between non-syndromic cleft lip and palate, and cancer HONGYI WANG, TAO QIU, JIE SHI, JIULONG LIANG, YANG WANG, LIANGLIANG QUAN, YU ZHANG, QIAN ZHANG and KAI TAO Department of Plastic Surgery, General Hospital of Shenyang Military Area Command, PLA, Shenyang, Liaoning 110016, P.R. China Received March 10, 2015; Accepted December 18, 2015 DOI: 10.3892/mmr.2016.4802 Abstract. The present study aimed to investigate the for NSCL/P were implicated predominantly in the TGF-β molecular mechanisms underlying non-syndromic cleft lip, signaling pathway, the cell cycle and in viral carcinogenesis. with or without cleft palate (NSCL/P), and the association The TP53, CDK1, SMAD3, PIK3R1 and CASP3 genes were between this disease and cancer. The GSE42589 data set found to be associated, not only with NSCL/P, but also with was downloaded from the Gene Expression Omnibus data- cancer. These results may contribute to a better understanding base, and contained seven dental pulp stem cell samples of the molecular mechanisms of NSCL/P. from children with NSCL/P in the exfoliation period, and six controls. Differentially expressed genes (DEGs) were Introduction screened using the RankProd method, and their potential functions were revealed by pathway enrichment analysis and Non-syndromic cleft lip, with or without cleft palate (NSCL/P) construction of a pathway interaction network. Subsequently, is one of the most common types of congenital defect and cancer genes were obtained from six cancer databases, and affects 3.4-22.9/10,000 individuals worldwide (1). -

Characterization of Budding Yeast Orc6 : a Dimerization Domain Is
Characterization of the role of Orc6 in the cell cycle of the budding yeast Saccharomyces cerevisiae by Jeffrey W. Semple A thesis presented to the University of Waterloo in fulfilment of the thesis requirement for the degree of Doctor of Philosophy in Biology Waterloo, Ontario, Canada, 2006 © Jeffrey W. Semple 2006 I hereby declare that I am the sole author of this thesis. This is a true copy of the thesis, including any required finalrevisions, as accepted by my examiners. I understand that my thesis may be made electronically available to the public. ii Abstract The heterohexameric origin recognition complex (ORC) acts as a scaffold for the G1 phase assembly of pre-replicative complexes. Only the Orc1-5 subunits are required for origin binding in budding yeast, yet Orc6 is an essential protein for cell proliferation. In comparison to other eukaryotic Orc6 proteins, budding yeast Orc6 appears to be quite divergent. Two-hybrid analysis revealed that Orc6 only weakly interacts with other ORC subunits. In this assay Orc6 showed a strong ability to self-associate, although the significance of this dimerization or multimerization remains unclear. Imaging of Orc6- eYFP revealed a punctate sub-nuclear localization pattern throughout the cell cycle, representing the first visualization of replication foci in live budding yeast cells. Orc6 was not detected at the site of division between mother and daughter cells, in contrast to observations from metazoans. An essential role for Orc6 in DNA replication was identified by depleting the protein before and during G1 phase. Surprisingly, Orc6 was required for entry into S phase after pre-replicative complex formation, in contrast to what has been observed for other ORC subunits.