Biallelic Variants in LAMB1 Causing Hydranencephaly: a Severe Phenotype of a Rare Malformative Encephalopathy
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Classification of Congenital Abnormalities of the CNS
315 Classification of Congenital Abnormalities of the CNS M. S. van der Knaap1 A classification of congenital cerebral, cerebellar, and spinal malformations is pre J . Valk2 sented with a view to its practical application in neuroradiology. The classification is based on the MR appearance of the morphologic abnormalities, arranged according to the embryologic time the derangement occurred. The normal embryology of the brain is briefly reviewed, and comments are made to explain the classification. MR images illustrating each subset of abnormalities are presented. During the last few years, MR imaging has proved to be a diagnostic tool of major importance in children with congenital malformations of the eNS [1]. The excellent gray fwhite-matter differentiation and multi planar imaging capabilities of MR allow a systematic analysis of the condition of the brain in infants and children. This is of interest for estimating prognosis and for genetic counseling. A classification is needed to serve as a guide to the great diversity of morphologic abnormalities and to make the acquired data useful. Such a system facilitates encoding, storage, and computer processing of data. We present a practical classification of congenital cerebral , cerebellar, and spinal malformations. Our classification is based on the morphologic abnormalities shown by MR and on the time at which the derangement of neural development occurred. A classification based on etiology is not as valuable because the various presumed causes rarely lead to a specific pattern of malformations. The abnor malities reflect the time the noxious agent interfered with neural development, rather than the nature of the noxious agent. The vulnerability of the various structures to adverse agents is greatest during the period of most active growth and development. -

Version 1.0, 8/21/2016 Zika Pregnancy Outcome Reporting Of
Version 1.0, 8/21/2016 Zika Pregnancy outcome reporting of brain abnormalities and other adverse outcomes The following box details the inclusion criteria for brain abnormalities and other adverse outcomes potentially related to Zika virus infection during pregnancy. All pregnancy outcomes are monitored, but weekly reporting of adverse outcomes is limited to those meeting the below criteria. All prenatal and postnatal adverse outcomes are reported for both Zika Pregnancy Registries (US Zika Pregnancy Registry, Zika Active Pregnancy Surveillance System) and Active Birth Defects Surveillance; however, case finding methods dictate some differences in specific case definitions. Brain abnormalities with and without microcephaly Confirmed or possible congenital microcephaly# Intracranial calcifications Cerebral atrophy Abnormal cortical formation (e.g., polymicrogyria, lissencephaly, pachygyria, schizencephaly, gray matter heterotopia) Corpus callosum abnormalities Cerebellar abnormalities Porencephaly Hydranencephaly Ventriculomegaly / hydrocephaly (excluding “mild” ventriculomegaly without other brain abnormalities) Fetal brain disruption sequence (collapsed skull, overlapping sutures, prominent occipital bone, scalp rugae) Other major brain abnormalities, including intraventricular hemorrhage in utero (excluding post‐natal IVH) Early brain malformations, eye abnormalities, or consequences of central nervous system (CNS) dysfunction Neural tube defects (NTD) o Anencephaly / Acrania o Encephalocele o Spina bifida Holoprosencephaly -
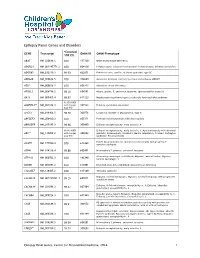
Epilepsy Panel Genes and Disorders
Epilepsy Panel Genes and Disorders *Covered GENE Transcript OMIM ID OMIM Phenotype 10X (%) ABAT NM_020686.5 100 137150 GABA-transaminase deficiency ADGRG1 NM_001145771.2 100 604110 Polymicrogyria, bilateral frontoparietal; Polymicrogyria, bilateral perisylvian ADGRV1 NM_032119.3 99.93 602851 Febrile seizures, familial, 4; Usher syndrome, type 2C ADRA2B NM_000682.5 100 104260 Autosomal dominant cortical myoclonus and epilepsy (ADCME) ADSL NM_000026.2 100 608222 Adenylosuccinase deficiency AFG3L2 NM_006796.2 98.16 604581 Ataxia, spastic, 5, autosomal recessive; Spinocerebellar ataxia 28 AKT3 NM_005465.4 99.97 611223 Megalencephaly-polymicrogyria-polydactyly-hydrocephalus syndrome 93.05 (100% ALDH7A1** NM_001182.3 with Sanger 107323 Epilepsy, pyridoxine-dependent Gap fill) ALG13 NM_018466.5 98.76 300776 Congenital disorder of glycosylation, type Is ARFGEF2 NM_006420.2 100 605371 Periventricular heterotopia with microcephaly ARHGEF9 NM_015185.3 99.96 300430 Epileptic encephalopathy, early infantile, 8 98.96 (100% Epileptic encephalopathy, early infantile, 1; Hydranencephaly with abnormal ARX** NM_139058.2 with Sanger 300382 genitalia; Lissencephaly, X-linked 2; Mental retardation, X-linked; Partington Gap fill) syndrome; Proud syndrome ASAH1 NM_177924.3 613468 Farber lipogranulomatosis; Spinal muscular atrophy with progressive 100 myoclonic epilepsy ASPM NM_018136.4 99.88 605481 Microcephaly 5, primary, autosomal recessive ATP1A2 NM_000702.3 182340 Alternating hemiplegia of childhood; Migraine, familial basilar; Migraine, 100 familial hemiplegic, -

Chapter III: Case Definition
NBDPN Guidelines for Conducting Birth Defects Surveillance rev. 06/04 Appendix 3.5 Case Inclusion Guidance for Potentially Zika-related Birth Defects Appendix 3.5 A3.5-1 Case Definition NBDPN Guidelines for Conducting Birth Defects Surveillance rev. 06/04 Appendix 3.5 Case Inclusion Guidance for Potentially Zika-related Birth Defects Contents Background ................................................................................................................................................. 1 Brain Abnormalities with and without Microcephaly ............................................................................. 2 Microcephaly ............................................................................................................................................................ 2 Intracranial Calcifications ......................................................................................................................................... 5 Cerebral / Cortical Atrophy ....................................................................................................................................... 7 Abnormal Cortical Gyral Patterns ............................................................................................................................. 9 Corpus Callosum Abnormalities ............................................................................................................................. 11 Cerebellar abnormalities ........................................................................................................................................ -

Blueprint Genetics Neuronal Migration Disorder Panel
Neuronal Migration Disorder Panel Test code: MA2601 Is a 59 gene panel that includes assessment of non-coding variants. Is ideal for patients with a clinical suspicion of neuronal migration disorder. About Neuronal Migration Disorder Neuronal migration disorders (NMDs) are a group of birth defects caused by the abnormal migration of neurons in the developing brain and nervous system. During development, neurons must migrate from the areas where they are originate to the areas where they will settle into their proper neural circuits. The structural abnormalities found in NMDs include schizencephaly, porencephaly, lissencephaly, agyria, macrogyria, polymicrogyria, pachygyria, microgyria, micropolygyria, neuronal heterotopias, agenesis of the corpus callosum, and agenesis of the cranial nerves. Mutations of many genes are involved in neuronal migration disorders, such as DCX in classical lissencephaly spectrum, TUBA1A in microlissencephaly with agenesis of the corpus callosum, and RELN and VLDLR in lissencephaly with cerebellar hypoplasia. Mutations in ARX cause a variety of phenotypes ranging from hydranencephaly or lissencephaly to early-onset epileptic encephalopathies, including Ohtahara syndrome and infantile spasms or intellectual disability with no brain malformations. Availability 4 weeks Gene Set Description Genes in the Neuronal Migration Disorder Panel and their clinical significance Gene Associated phenotypes Inheritance ClinVar HGMD ACTB* Baraitser-Winter syndrome AD 55 60 ACTG1* Deafness, Baraitser-Winter syndrome AD 27 47 ADGRG1 -

Hydranencephaly
Kathmandu University Medical Journal (2010), Vol. 8, No. 1, Issue 29, 83-86 Case Note Hydranencephaly Pant S1, Kaur G2, JK De3 1Medical Offi cer, 2Associate Professor, 3Professor and Head, Department of Obstetrics and Gynaecology, Manipal College of Medical Sciences Abstract Hydranencephaly is a rare congenital condition where the greater portions of the cerebral hemispheres and the corpus striatum are replaced by cerebrospinal fl uid and glial tissue. The meninges and the skull are well formed, which is consistent with earlier normal embryogenesis of the telencephalon. Bilateral occlusion of the internal carotid arteries in utero is a potential mechanism. Clinical features include intact brainstem refl exes without evidence of higher cortical activity. The infant’s head size and the spontaneous refl exes such as sucking, swallowing, crying, and moving the arms and legs may all seem normal at birth. However, after a few weeks the infant usually becomes irritable and has increased muscle tone and after a few months of life, seizures and hydrocephalus (excessive accumulation of cerebrospinal fl uid in the brain) may develop. Other symptoms may include visual impairment, lack of growth, deafness, blindness, spastic quadriparesis (paralysis), and intellectual defi cits. Since the early behaviour appears to be relatively normal, the diagnosis may be delayed for months sometimes. There is no defi nitive treatment for hydranencephaly. The outlook for children with hydranencephaly is generally poor, and many children with this disorder die before their fi rst birthday. Key words: hydranencephaly, congenital anomaly, vascular disruption, thromboplastin, 19 years old, Gravida 2 Para 0 Abortion 1, presented HBV, blood sugars) were normal. -

Hydranencephaly in Association with Roberts Syndrome
LE JOURNAL CANAD1EN DES SCIENCES NEUROLOGIQUKS Hydranencephaly in Association with Roberts Syndrome CHRIS E. U. EKONG AND BOHDAN ROZDILSKY SUMMARY: A clinicopathological In 1919, Dr. John Roberts de perforate. Marked hypertelorism study in a case of Roberts syndrome (tet- scribed a brother and sister with tet- was noted. The head appeared nor raphocomelia, cleft lip and palate, and raphocomelia, cleft lip and palate, mal in size but the neck was short and phallic hypertrophy) is reported. This pa and phallic hypertrophy. This com stiff. The infant died a few minutes tient had hydranencephaly and imperfo bination of congenital abnormalities after birth. Family history revealed rate anus, two additional congenital ab has subsequently been named that the patient's mother's two sis normalities so far not reported in this ters had no children, as they spon syndrome. Roberts syndrome. Twenty-two similar cases have been reported taneously aborted during each preg RESUME: Nous rapportons Vetude (Appelt et al., 1966; Holmes et al., nancy. The patient's parents were clinico pathologique d'un cas de syn 1972; Freeman et al., 1974). not consanguineously related. drome de Roberts (tetraphocomelie, bee Other congenital anomalies in A post mortem total body radio de lievre et hypertrophic phallique). Ce clude cryptorchidism, patent ductus graph (Figure 2) revealed ox patient presentait egalement une arteriosus and foramen ovale, en- ycephalic calvarium. The tubular hydranencephalie et tin anus non perfore cephalocele, polycystic kidneys, bones of the upper and lower ex — deux anomalies congenitales non horse-shoe kidneys and deformity of tremities were moderately well de prealablement connues dans ce syn base of skull and cribriform plate veloped, but were short compared to drome. -

The Expanding Phenotype of COL4A1 and COL4A2 Mutations: Clinical Data on 13 Newly Identified Families and a Review of the Literature
© American College of Medical Genetics and Genomics REVIEW The expanding phenotype of COL4A1 and COL4A2 mutations: clinical data on 13 newly identified families and a review of the literature Marije E.C. Meuwissen, MD, PhD1,2, Dicky J.J. Halley, PhD1, Liesbeth S. Smit, MD3, Maarten H. Lequin, MD, PhD4, Jan M. Cobben, MD, PhD5, René de Coo, MD, PhD3, Jeske van Harssel, MD6, Suzanne Sallevelt, MD7, Gwendolyn Woldringh, MD, PhD8, Marjo S. van der Knaap, MD, PhD9, Linda S. de Vries, MD, PhD10 and Grazia M.S. Mancini, MD, PhD1 Two proα1(IV) chains, encoded by COL4A1, form trimers that children with porencephaly or other patterns of parenchymal hemor- contain, in addition, a proα2(IV) chain encoded by COL4A2 and rhage, with a high de novo mutation rate of 40% (10/24). The obser- are the major component of the basement membrane in many tis- vations in 13 novel families harboring either COL4A1 or COL4A2 sues. Since 2005, COL4A1 mutations have been known as an auto- mutations prompted us to review the clinical spectrum. We observed somal dominant cause of hereditary porencephaly. COL4A1 and recognizable phenotypic patterns and propose a screening protocol COL4A2 mutations have been reported with a broader spectrum at diagnosis. Our data underscore the importance of COL4A1 and of cerebrovascular, renal, ophthalmological, cardiac, and muscular COL4A2 mutations in cerebrovascular disease, also in sporadic abnormalities, indicated as “COL4A1 mutation–related disorders.” patients. Follow-up data on symptomatic and asymptomatic muta- Genetic counseling is challenging because of broad phenotypic varia- tion carriers are needed for prognosis and appropriate surveillance. -

Hydranencephaly
Pavone et al. Italian Journal of Pediatrics 2014, 40:79 http://www.ijponline.net/content/40/1/79 ITALIAN JOURNAL OF PEDIATRICS REVIEW Open Access Hydranencephaly: cerebral spinal fluid instead of cerebral mantles Piero Pavone1*, Andrea D Praticò1, Giovanna Vitaliti1, Martino Ruggieri2, Renata Rizzo3, Enrico Parano4, Lorenzo Pavone1, Giuseppe Pero5 and Raffaele Falsaperla1 Abstract The authors report a wide and updated revision of hydranencephaly, including a literature review, and present the case of a patient affected by this condition, still alive at 36 months. Hydranencephaly is an isolated and with a severe prognosis abnormality, affecting the cerebral mantle. In this condition, the cerebral hemispheres are completely or almost completely absent and are replaced by a membranous sac filled with cerebrospinal fluid. Midbrain is usually not involved. Hydranencephaly is a relatively rare cerebral disorder. Differential diagnosis is mainly relevant when considering severe hydrocephalus, poroencephalic cyst and alobar holoprosencephaly. Ethical questions related to the correct criteria for the surgical treatment are also discussed. Keywords: Hydranencephaly, Holoprosencephaly, Congenital anomaly, Brain malformation, Severe hydrocephalus Introduction often misdiagnosed due to the similarities of HE to disor- Hydranencephaly (HE) is a rare, mostly isolated abnormal- ders involving the cerebral mantle (Table 1). ity, which is reported to affect about 1 out 5000 continu- The aim of our review is to report on this condition, ing pregnancies [1,2]; an accurate incidence is difficult to utilizing data from the literature, and also presenting our determine, considering how similar this condition is to experience with a patient affected by the bilateral form of others and the limited diagnostic techniques that have HE, who is still alive at the age of 36 months. -

State of the Art &&&&&&&&&&&&&& Neuroimaging and the Timing of Fetal and Neonatal Brain Injury
State of the Art &&&&&&&&&&&&&& Neuroimaging and the Timing of Fetal and Neonatal Brain Injury Patrick D. Barnes, MD modalities provide spatial resolution based upon physiologic or metabolic data. Some modalities may actually be considered to provide both structural and functional information. Ultrasonography (US) Current and advanced structural and functional neuroimaging techniques is primarily a structural imaging modality with some functional 1±11 are presented along with guidelines for utilization and principles of imaging capabilities (e.g., Doppler ÐFigure 1). It is readily accessible, diagnosis in fetal and neonatal central nervous system abnormalities. portable, fast, real time and multiplanar. It less expensive than other Pattern of injury, timing issues, and differential diagnosis are addressed with cross-sectional modalities and relatively noninvasive (nonionizing emphasis on neurovascular disease. Ultrasonography and computed radiation). It requires no contrast agent and infrequently needs tomography provide relatively rapid and important screening information patient sedation. The resolving power of US is based on variations in regarding gross macrostructural abnormalities. However, current and acoustic reflectance of tissues. Its diagnostic effectiveness, however, is advanced MRI techniques often provide more definitive macrostructural, primarily dependent upon the skill and experience of the operator and microstructural, and functional imaging information. interpreter. Also, US requires a window or path unimpeded by bone -

Bilateral Schizencephaly with Septo Optic Dysplasia Rare Cause of Seizure Disorder
Faridpur Med. Coll. J. 2018;13(1):50-52 Case Report Bilateral Schizencephaly with Septo Optic Dysplasia Rare Cause of Seizure Disorder MMB Zaman Abstract: Schizencephaly is an extremely rare developmental birth defect characterized by abnormal slits or clefts in the cerebral hemispheres extending from the lateral ventricle to the cerebral cortex. The margins of the cleft are lined with heterotropic, dysplastic gray matter. The causes of schizencephaly are heterogenous and include teratogens, prenatal infarction/infections, maternal trauma, or EMX2 mutations. It is a central nervous system disorder with variable presentations. People with this disorder commonly have developmental delays, delays in speech and language skills, seizures disorder and problems with brain-spinal cord communication. This condition is present at birth and manifests early in life. This patient presented with seizure and growth retardation and investigation revealed bilateral Schizencephaly with Septo optic dysplasia. Key words: Schizencephaly, Clefts, Septo Optic Dysplasia. Introduction: Schizencephaly is a rare cortical malformation that Other factors such as infection, metabolic disorders, manifests as a gray matter-lined cleft extending from and genetic defects also play an role in the the ependyma to the pia mater. In 1946, Yakovlev and development of schizencephaly. Granata T et al. have Wadsworth first described schizencephaly as reported heterozygous mutations of the EMX2 gene hemispheric clefts in the region of the primary fissures, associated with schizencephaly3. infolding of gray matter along the clefts, and associated cerebral malformations. The schizencephaly clefts are Since the exact cause of the disorder is unknown, it's mostly perisylvian or centrally located1. hard to pinpoint risk factors. -

Typical Lesions in the Fetal Nervous System: Correlations Between Fetal Magnetic Resonance Imaging and Obstetric Ultrasonography Findings
Typical lesions in the fetal nervous system: correlations between fetal magnetic resonance imaging and obstetric ultrasonography findings PICTORIAL ESSAY Heron Werner1, Taisa Davaus Gasparetto1, Pedro Daltro1, Emerson Leandro Gasparetto1, https://doi.org/10.14366/usg.17040 2 Edward Araujo Júnior pISSN: 2288-5919 • eISSN: 2288-5943 Ultrasonography 2018;37:261-274 1Department of Radiology, Clínica de Diagnóstico por Imagem (CDPI), Rio de Janeiro; 2Department of Obstetrics, Paulista School of Medicine, Federal University of São Paulo (EPM-UNIFESP), São Paulo, Brazil Received: May 29, 2017 Revised: October 21, 2017 Central nervous system (CNS) malformations play a role in all fetal malformations. Accepted: October 21, 2017 Ultrasonography (US) is the best screening method for identifying fetal CNS malformations. Correspondence to: Edward Araujo Júnior, PhD, Department A good echographic study depends on several factors, such as positioning, fetal mobility and of Obstetrics, Paulista School of growth, the volume of amniotic fluid, the position of the placenta, the maternal wall, the quality Medicine, Federal University of São Paulo (EPM-UNIFESP), Rua Belchior de of the apparatus, and the sonographer’s experience. Although US is the modality of choice for Azevedo, 156 apto. 111 Torre Vitoria, routine prenatal follow-up because of its low cost, wide availability, safety, good sensitivity, São Paulo, CEP 05089-030, Brazil and real-time capability, magnetic resonance imaging (MRI) is promising for the morphological Tel. +55-11-37965944 Fax. +55-11-37965944 evaluation of fetuses that otherwise would not be appropriately evaluated using US. The aim of E-mail: [email protected] this article is to present correlations of fetal MRI findings with US findings for the major CNS malformations.