Congenital Anomalies of the Brain and Spinal Cord Edgar A
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Catastrophic Intramedullary Abscess Caused by a Missed Congenital Dermal Sinus
online © ML Comm www.jkns.or.kr http://dx.doi.org/10.3340/jkns.2015.57.3.225 Print ISSN 2005-3711 On-line ISSN 1598-7876 J Korean Neurosurg Soc 57 (3) : 225-228, 2015 Copyright © 2015 The Korean Neurosurgical Society Case Report Catastrophic Intramedullary Abscess Caused by a Missed Congenital Dermal Sinus Yun-Sik Dho, M.D., Seung-Ki Kim, M.D., Ph.D., Kyu-Chang Wang, M.D., Ph.D., Ji Hoon Phi, M.D. Division of Pediatric Neurosurgery, Seoul National University Children’s Hospital, Seoul National University College of Medicine, Seoul, Korea Congenital dermal sinus (CDS) is a type of occult spinal dysraphism characterized by a midline skin dimple. A 12-month-old girl presented with fe- ver and ascending quadriparesis. She had a midline skin dimple in the upper sacral area that had been discovered in her neonatal period. Imaging studies revealed a holocord intramedullary abscess and CDS. Overlooking CDS or misdiagnosing it as benign sacrococcygeal dimple may lead to catastrophic infection and cause serious neurological deficits. Therefore, further imaging work-up or consultation with a pediatric neurosurgeon is recommended following discovery of any atypical-looking dimples in the midline. Key Words : Congenital dermal sinus · Intramedullary abscess · Diagnosis. INTRODUCTION into the spinal canal. Unfortunately, a detailed history report re- vealed that the skin ostium was detected early in her neonatal Congenital dermal sinus (CDS) is a congenital anomaly that period by the parents, but they thought it was a benign skin le- develops anywhere in the midline along the neuraxis1). CDS be- sion with little clinical importance. -

Classification of Congenital Abnormalities of the CNS
315 Classification of Congenital Abnormalities of the CNS M. S. van der Knaap1 A classification of congenital cerebral, cerebellar, and spinal malformations is pre J . Valk2 sented with a view to its practical application in neuroradiology. The classification is based on the MR appearance of the morphologic abnormalities, arranged according to the embryologic time the derangement occurred. The normal embryology of the brain is briefly reviewed, and comments are made to explain the classification. MR images illustrating each subset of abnormalities are presented. During the last few years, MR imaging has proved to be a diagnostic tool of major importance in children with congenital malformations of the eNS [1]. The excellent gray fwhite-matter differentiation and multi planar imaging capabilities of MR allow a systematic analysis of the condition of the brain in infants and children. This is of interest for estimating prognosis and for genetic counseling. A classification is needed to serve as a guide to the great diversity of morphologic abnormalities and to make the acquired data useful. Such a system facilitates encoding, storage, and computer processing of data. We present a practical classification of congenital cerebral , cerebellar, and spinal malformations. Our classification is based on the morphologic abnormalities shown by MR and on the time at which the derangement of neural development occurred. A classification based on etiology is not as valuable because the various presumed causes rarely lead to a specific pattern of malformations. The abnor malities reflect the time the noxious agent interfered with neural development, rather than the nature of the noxious agent. The vulnerability of the various structures to adverse agents is greatest during the period of most active growth and development. -

Closed Spinal Dysraphism and Tethered Cord
ACNRSO14_Layout 1 04/09/2014 22:14 Page 28 NEUROSURGERY ARTICLE Ruth-Mary deSouza trained in medicine at Closed Spinal Dysraphism Guy’s, Kings and St Thomas Medical School and graduated in 2008. She entered the London and Tethered Cord Neurosurgery training programme in 2010 and is currently an ST5 trainee on the South Thames Syndrome: A Review of Neurosurgery programme. David Frim Multidisciplinary Team Management is Professor of Surgery, Neurology and Paediatrics at the University of Chicago. He is an Summary internationally recognised • Embryology of spinal dysraphism clinical Neurosurgeon and Neurosciences Researcher • Clinical features of tethered cord syndrome who specialises in the care • Multidisciplinary management of closed spinal dysraphism of children and adults with congenital neurosurgical problems. Currently, Dr Frim serves as principal investigator on laboratory studies related to neural injury and clinical studies focusing on Abstract outcomes after treatment of congenital anomalies of the nervous system especially as related to cognition. The initial diagnosis as well as the long term management of occult spinal dysraphism and Dr Frim is joint senior author of the article. tethered spinal cord is often managed by a large number of healthcare professionals including Paediatricians, GPs, Neurologists, Neurosurgeons, Rehabilitation Physicians and Paige Terrien Church Therapists. We review the entity of spinal dysraphism. An approach to the evaluation and is an Assistant Professor of diagnosis of these entities is subsequently discussed. In addition, concepts involved in the Paediatrics at the pathophysiology, neurosurgical repair, and outcome are presented in the context of postop - University of Toronto. She is the Director of the erative management issues that rely upon the knowledge of all professionals who may Neonatal Follow Up Clinic encounter these patients. -

Version 1.0, 8/21/2016 Zika Pregnancy Outcome Reporting Of
Version 1.0, 8/21/2016 Zika Pregnancy outcome reporting of brain abnormalities and other adverse outcomes The following box details the inclusion criteria for brain abnormalities and other adverse outcomes potentially related to Zika virus infection during pregnancy. All pregnancy outcomes are monitored, but weekly reporting of adverse outcomes is limited to those meeting the below criteria. All prenatal and postnatal adverse outcomes are reported for both Zika Pregnancy Registries (US Zika Pregnancy Registry, Zika Active Pregnancy Surveillance System) and Active Birth Defects Surveillance; however, case finding methods dictate some differences in specific case definitions. Brain abnormalities with and without microcephaly Confirmed or possible congenital microcephaly# Intracranial calcifications Cerebral atrophy Abnormal cortical formation (e.g., polymicrogyria, lissencephaly, pachygyria, schizencephaly, gray matter heterotopia) Corpus callosum abnormalities Cerebellar abnormalities Porencephaly Hydranencephaly Ventriculomegaly / hydrocephaly (excluding “mild” ventriculomegaly without other brain abnormalities) Fetal brain disruption sequence (collapsed skull, overlapping sutures, prominent occipital bone, scalp rugae) Other major brain abnormalities, including intraventricular hemorrhage in utero (excluding post‐natal IVH) Early brain malformations, eye abnormalities, or consequences of central nervous system (CNS) dysfunction Neural tube defects (NTD) o Anencephaly / Acrania o Encephalocele o Spina bifida Holoprosencephaly -
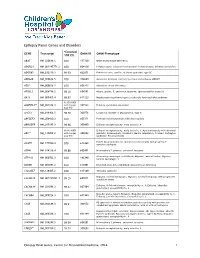
Epilepsy Panel Genes and Disorders
Epilepsy Panel Genes and Disorders *Covered GENE Transcript OMIM ID OMIM Phenotype 10X (%) ABAT NM_020686.5 100 137150 GABA-transaminase deficiency ADGRG1 NM_001145771.2 100 604110 Polymicrogyria, bilateral frontoparietal; Polymicrogyria, bilateral perisylvian ADGRV1 NM_032119.3 99.93 602851 Febrile seizures, familial, 4; Usher syndrome, type 2C ADRA2B NM_000682.5 100 104260 Autosomal dominant cortical myoclonus and epilepsy (ADCME) ADSL NM_000026.2 100 608222 Adenylosuccinase deficiency AFG3L2 NM_006796.2 98.16 604581 Ataxia, spastic, 5, autosomal recessive; Spinocerebellar ataxia 28 AKT3 NM_005465.4 99.97 611223 Megalencephaly-polymicrogyria-polydactyly-hydrocephalus syndrome 93.05 (100% ALDH7A1** NM_001182.3 with Sanger 107323 Epilepsy, pyridoxine-dependent Gap fill) ALG13 NM_018466.5 98.76 300776 Congenital disorder of glycosylation, type Is ARFGEF2 NM_006420.2 100 605371 Periventricular heterotopia with microcephaly ARHGEF9 NM_015185.3 99.96 300430 Epileptic encephalopathy, early infantile, 8 98.96 (100% Epileptic encephalopathy, early infantile, 1; Hydranencephaly with abnormal ARX** NM_139058.2 with Sanger 300382 genitalia; Lissencephaly, X-linked 2; Mental retardation, X-linked; Partington Gap fill) syndrome; Proud syndrome ASAH1 NM_177924.3 613468 Farber lipogranulomatosis; Spinal muscular atrophy with progressive 100 myoclonic epilepsy ASPM NM_018136.4 99.88 605481 Microcephaly 5, primary, autosomal recessive ATP1A2 NM_000702.3 182340 Alternating hemiplegia of childhood; Migraine, familial basilar; Migraine, 100 familial hemiplegic, -

Anesthetic Management of a Neonate with Antley-Bixler Syndrome -A Case Report
Anesth Pain Med 2011; 6: 89~92 ■Case Report■ Anesthetic management of a neonate with Antley-Bixler syndrome -A case report- Department of Anesthesiology and Pain Medicine, Sanggye Paik Hospital, Inje University College of Medicine, Seoul, Korea Young-Suk Kwon, Jae Keun Jo, Yun-Hee Lim, Jun Heum Yon, and Kye-Min Kim Antley-Bixler syndrome is a congenital anomaly of multiple bones has been reported to be approximately 45−50% [2]; and cartilage, and this was first reported by Antley and Bixler in considering the potential risk of respiratory complications in 1975. It is characterized by craniosynostosis, midface hypoplasia this syndrome, anesthesiologists should be vigilant during with choanal stenosis and atresia, radiohumeral synostosis and femoral bowing. This is sometimes accompanied by cardiac, renal, general anesthesia, especially in terms of airway management. gastrointestinal and genital malformations. The risk of respiratory In addition, careful attention is needed during patient posi- distress is high in the infants with this syndrome, and this is most tioning under anesthesia to prevent accidental injuries. commonly caused by choanal stenosis and atresia. Careful anes- thetic management is needed for these infants because of the Here we report a case of general anesthesia for a neonate potential risk of a difficult airway and respiratory complications. We with ABS who underwent neurosurgical operations for the report here on our experience with the anesthetic management of correction of congenital dermal sinus and hydrocephalus. a neonate with Antley-Bixler syndrome and we review the relevant literature. (Anesth Pain Med 2011; 6: 89∼92) CASE REPORT Key Words: Anesthesia, Antley-Bixler syndrome, Craniosyno- stosis. -

Chapter III: Case Definition
NBDPN Guidelines for Conducting Birth Defects Surveillance rev. 06/04 Appendix 3.5 Case Inclusion Guidance for Potentially Zika-related Birth Defects Appendix 3.5 A3.5-1 Case Definition NBDPN Guidelines for Conducting Birth Defects Surveillance rev. 06/04 Appendix 3.5 Case Inclusion Guidance for Potentially Zika-related Birth Defects Contents Background ................................................................................................................................................. 1 Brain Abnormalities with and without Microcephaly ............................................................................. 2 Microcephaly ............................................................................................................................................................ 2 Intracranial Calcifications ......................................................................................................................................... 5 Cerebral / Cortical Atrophy ....................................................................................................................................... 7 Abnormal Cortical Gyral Patterns ............................................................................................................................. 9 Corpus Callosum Abnormalities ............................................................................................................................. 11 Cerebellar abnormalities ........................................................................................................................................ -

Statistical Analysis Plan
Cover Page for Statistical Analysis Plan Sponsor name: Novo Nordisk A/S NCT number NCT03061214 Sponsor trial ID: NN9535-4114 Official title of study: SUSTAINTM CHINA - Efficacy and safety of semaglutide once-weekly versus sitagliptin once-daily as add-on to metformin in subjects with type 2 diabetes Document date: 22 August 2019 Semaglutide s.c (Ozempic®) Date: 22 August 2019 Novo Nordisk Trial ID: NN9535-4114 Version: 1.0 CONFIDENTIAL Clinical Trial Report Status: Final Appendix 16.1.9 16.1.9 Documentation of statistical methods List of contents Statistical analysis plan...................................................................................................................... /LQN Statistical documentation................................................................................................................... /LQN Redacted VWDWLVWLFDODQDO\VLVSODQ Includes redaction of personal identifiable information only. Statistical Analysis Plan Date: 28 May 2019 Novo Nordisk Trial ID: NN9535-4114 Version: 1.0 CONFIDENTIAL UTN:U1111-1149-0432 Status: Final EudraCT No.:NA Page: 1 of 30 Statistical Analysis Plan Trial ID: NN9535-4114 Efficacy and safety of semaglutide once-weekly versus sitagliptin once-daily as add-on to metformin in subjects with type 2 diabetes Author Biostatistics Semaglutide s.c. This confidential document is the property of Novo Nordisk. No unpublished information contained herein may be disclosed without prior written approval from Novo Nordisk. Access to this document must be restricted to relevant parties.This -

Blueprint Genetics Neuronal Migration Disorder Panel
Neuronal Migration Disorder Panel Test code: MA2601 Is a 59 gene panel that includes assessment of non-coding variants. Is ideal for patients with a clinical suspicion of neuronal migration disorder. About Neuronal Migration Disorder Neuronal migration disorders (NMDs) are a group of birth defects caused by the abnormal migration of neurons in the developing brain and nervous system. During development, neurons must migrate from the areas where they are originate to the areas where they will settle into their proper neural circuits. The structural abnormalities found in NMDs include schizencephaly, porencephaly, lissencephaly, agyria, macrogyria, polymicrogyria, pachygyria, microgyria, micropolygyria, neuronal heterotopias, agenesis of the corpus callosum, and agenesis of the cranial nerves. Mutations of many genes are involved in neuronal migration disorders, such as DCX in classical lissencephaly spectrum, TUBA1A in microlissencephaly with agenesis of the corpus callosum, and RELN and VLDLR in lissencephaly with cerebellar hypoplasia. Mutations in ARX cause a variety of phenotypes ranging from hydranencephaly or lissencephaly to early-onset epileptic encephalopathies, including Ohtahara syndrome and infantile spasms or intellectual disability with no brain malformations. Availability 4 weeks Gene Set Description Genes in the Neuronal Migration Disorder Panel and their clinical significance Gene Associated phenotypes Inheritance ClinVar HGMD ACTB* Baraitser-Winter syndrome AD 55 60 ACTG1* Deafness, Baraitser-Winter syndrome AD 27 47 ADGRG1 -

Principles of Pediatric Neurosurgery
Principles of Pediatric Neurosurgery Series Editor: Anthony J. Raimondi Principles of Pediatric Neurosurgery Head Injuries in the Newborn and Infant The Pediatric Spine 1: Development and Dysraphic State The Pediatric Spine II: Developmental Anomalies The Pediatric Spine III: Cysts, Tumors, and Infections Edited by Anthony J. Raimondi, Maurice Choux, and Concezio Di Rocco The Pediatric Spine III Cysts, Tumors, and Infections Edited by Anthony J. Raimondi, Maurice Choux, and Concezio Di Rocco With 87 Figures Springer Verlag New York Berlin Heidelberg London Paris Tokyo ANTHONY J. RAIMONDI, 37020 Gargagnago (Verona), Italy MAURICE CHOUX, M.D., H6pital des Enfants de la Timone, Rue Saint Pierre, 13005 Marseille, France CONCEZIO 01 Rocco, M.D., Istituto di Neurochirurgia, Universita Cattolica del Sacro Cuore, Largo Gemelli 8, 00168 Rome, Italy Library of Congress Cataloging-in-Publication Data The pediatric spine. (Principles of pediatric neurosurgery) Includes bibliographies and index. Contents: I. Development and the dysraphic state- - 3. Cysts, tumors, and infections. I. Spine-Diseases. 2. Spine-Abnormalities. 3. Spine-Surgery. 4. Pediatric neurology. I. Raimondi, Anthony J., 1928- . II. Choux, M. (Maurice) III. Di Rocco, C. (Concezio) IV. Series. [DNLM: I. Spinal Diseases-in infancy and childhood. 2. Spine growth & development. WE 725 P3711] RD768.P36 1989 618.92'73 88-24822 © 1989 by Springer-Verlag New York Inc. Softcover reprint of the hardcover 1st edition 1989 All rights reserved. This work may not be translated or copied in whole or in part without the written permission of the publisher (Springer-Verlag, 175 Fifth Avenue, New York, NY 10010, USA), except for brief excerpts in connection with reviews or scholarly analysis. -

Hydranencephaly
Kathmandu University Medical Journal (2010), Vol. 8, No. 1, Issue 29, 83-86 Case Note Hydranencephaly Pant S1, Kaur G2, JK De3 1Medical Offi cer, 2Associate Professor, 3Professor and Head, Department of Obstetrics and Gynaecology, Manipal College of Medical Sciences Abstract Hydranencephaly is a rare congenital condition where the greater portions of the cerebral hemispheres and the corpus striatum are replaced by cerebrospinal fl uid and glial tissue. The meninges and the skull are well formed, which is consistent with earlier normal embryogenesis of the telencephalon. Bilateral occlusion of the internal carotid arteries in utero is a potential mechanism. Clinical features include intact brainstem refl exes without evidence of higher cortical activity. The infant’s head size and the spontaneous refl exes such as sucking, swallowing, crying, and moving the arms and legs may all seem normal at birth. However, after a few weeks the infant usually becomes irritable and has increased muscle tone and after a few months of life, seizures and hydrocephalus (excessive accumulation of cerebrospinal fl uid in the brain) may develop. Other symptoms may include visual impairment, lack of growth, deafness, blindness, spastic quadriparesis (paralysis), and intellectual defi cits. Since the early behaviour appears to be relatively normal, the diagnosis may be delayed for months sometimes. There is no defi nitive treatment for hydranencephaly. The outlook for children with hydranencephaly is generally poor, and many children with this disorder die before their fi rst birthday. Key words: hydranencephaly, congenital anomaly, vascular disruption, thromboplastin, 19 years old, Gravida 2 Para 0 Abortion 1, presented HBV, blood sugars) were normal. -

Hydranencephaly in Association with Roberts Syndrome
LE JOURNAL CANAD1EN DES SCIENCES NEUROLOGIQUKS Hydranencephaly in Association with Roberts Syndrome CHRIS E. U. EKONG AND BOHDAN ROZDILSKY SUMMARY: A clinicopathological In 1919, Dr. John Roberts de perforate. Marked hypertelorism study in a case of Roberts syndrome (tet- scribed a brother and sister with tet- was noted. The head appeared nor raphocomelia, cleft lip and palate, and raphocomelia, cleft lip and palate, mal in size but the neck was short and phallic hypertrophy) is reported. This pa and phallic hypertrophy. This com stiff. The infant died a few minutes tient had hydranencephaly and imperfo bination of congenital abnormalities after birth. Family history revealed rate anus, two additional congenital ab has subsequently been named that the patient's mother's two sis normalities so far not reported in this ters had no children, as they spon syndrome. Roberts syndrome. Twenty-two similar cases have been reported taneously aborted during each preg RESUME: Nous rapportons Vetude (Appelt et al., 1966; Holmes et al., nancy. The patient's parents were clinico pathologique d'un cas de syn 1972; Freeman et al., 1974). not consanguineously related. drome de Roberts (tetraphocomelie, bee Other congenital anomalies in A post mortem total body radio de lievre et hypertrophic phallique). Ce clude cryptorchidism, patent ductus graph (Figure 2) revealed ox patient presentait egalement une arteriosus and foramen ovale, en- ycephalic calvarium. The tubular hydranencephalie et tin anus non perfore cephalocele, polycystic kidneys, bones of the upper and lower ex — deux anomalies congenitales non horse-shoe kidneys and deformity of tremities were moderately well de prealablement connues dans ce syn base of skull and cribriform plate veloped, but were short compared to drome.