Hydranencephaly
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Anencephaly
Glossary of Birth Anomaly Terms: A Anencephaly: A deadly birth anomaly where most of the brain and skull did not form. Anomaly: Any part of the body or chromosomes that has an unusual or irregular structure. Aortic valve stenosis: The aortic valve controls the flow of blood from the left ventricle of the heart to the aorta, which takes the blood to the rest of the body. If there is stenosis of this valve, the valve has space for blood to flow through, but it is too narrow. Atresia: Lack of an opening where there should be one. Atrial septal defect: An opening in the wall (septum) that separates the left and right top chambers (atria) of the heart. A hole can vary in size and may close on its own or may require surgery. Atrioventricular septal defect (endocardial cushion defect): A defect in both the lower portion of the atrial septum and the upper portion of the ventricular septum. Together, these defects make a large opening (canal) in the middle part of the heart. Aniridia (an-i-rid-e-a): An eye anomaly where the colored part of the eye (called the iris) is partly or totally missing. It usually affects both eyes. Other parts of the eye can also be formed incorrectly. The effects on children’s ability to see can range from mild problems to blindness. To learn more about aniridia, go to the U.S. National Library of Medicine website. Anophthalmia/microphthalmia (an-oph-thal-mia/mi-croph-thal-mia): Birth anomalies of the eyes. In anophthalmia, a baby is born without one or both eyes. -

Classification of Congenital Abnormalities of the CNS
315 Classification of Congenital Abnormalities of the CNS M. S. van der Knaap1 A classification of congenital cerebral, cerebellar, and spinal malformations is pre J . Valk2 sented with a view to its practical application in neuroradiology. The classification is based on the MR appearance of the morphologic abnormalities, arranged according to the embryologic time the derangement occurred. The normal embryology of the brain is briefly reviewed, and comments are made to explain the classification. MR images illustrating each subset of abnormalities are presented. During the last few years, MR imaging has proved to be a diagnostic tool of major importance in children with congenital malformations of the eNS [1]. The excellent gray fwhite-matter differentiation and multi planar imaging capabilities of MR allow a systematic analysis of the condition of the brain in infants and children. This is of interest for estimating prognosis and for genetic counseling. A classification is needed to serve as a guide to the great diversity of morphologic abnormalities and to make the acquired data useful. Such a system facilitates encoding, storage, and computer processing of data. We present a practical classification of congenital cerebral , cerebellar, and spinal malformations. Our classification is based on the morphologic abnormalities shown by MR and on the time at which the derangement of neural development occurred. A classification based on etiology is not as valuable because the various presumed causes rarely lead to a specific pattern of malformations. The abnor malities reflect the time the noxious agent interfered with neural development, rather than the nature of the noxious agent. The vulnerability of the various structures to adverse agents is greatest during the period of most active growth and development. -

Supratentorial Brain Malformations
Supratentorial Brain Malformations Edward Yang, MD PhD Department of Radiology Boston Children’s Hospital 1 May 2015/ SPR 2015 Disclosures: Consultant, Corticometrics LLC Objectives 1) Review major steps in the morphogenesis of the supratentorial brain. 2) Categorize patterns of malformation that result from failure in these steps. 3) Discuss particular imaging features that assist in recognition of these malformations. 4) Reference some of the genetic bases for these malformations to be discussed in greater detail later in the session. Overview I. Schematic overview of brain development II. Abnormalities of hemispheric cleavage III. Commissural (Callosal) abnormalities IV. Migrational abnormalities - Gray matter heterotopia - Pachygyria/Lissencephaly - Focal cortical dysplasia - Transpial migration - Polymicrogyria V. Global abnormalities in size (proliferation) VI. Fetal Life and Myelination Considerations I. Schematic Overview of Brain Development Embryology Top Mid-sagittal Top Mid-sagittal Closed Neural Tube (4 weeks) Corpus Callosum Callosum Formation Genu ! Splenium Cerebral Hemisphere (11-20 weeks) Hemispheric Cleavage (4-6 weeks) Neuronal Migration Ventricular/Subventricular Zones Ventricle ! Cortex (8-24 weeks) Neuronal Precursor Generation (Proliferation) (6-16 weeks) Embryology From ten Donkelaar Clinical Neuroembryology 2010 4mo 6mo 8mo term II. Abnormalities of Hemispheric Cleavage Holoprosencephaly (HPE) Top Mid-sagittal Imaging features: Incomplete hemispheric separation + 1)1) No septum pellucidum in any HPEs Closed Neural -

Prenatal Diagnosis, Phenotypic and Obstetric Characteristics of Holoprosencephaly
Fetal Diagn Ther 2005;20:161–166 Received: August 6, 2003 DOI: 10.1159/000083897 Accepted after revision: January 15, 2004 Prenatal Diagnosis, Phenotypic and Obstetric Characteristics of Holoprosencephaly Ga´ bor J. Joo´ Artu´ r Beke Csaba Papp Erno˝To´ th-Pa´ l Zsanett Szigeti Zolta´nBa´ n Zolta´ n Papp Semmelweis University Medical School I., Department of Obstetrics and Gynecology, Budapest, Hungary Key Words Introduction Holoprosencephaly W Prenataldiagnosis W Patient’s history W Accompanying malformations In the early weeks of embryonic development, the brain consists of three parts: prosencephalon, mesenceph- alon, rhombencephalon. Between the time of neural tube Abstract closure and the 5th gestational week, the prosencephalon The diagnosis of fetal malformations, especially those of gives origin to telencephalon (cerebral hemispheres) and the central nervous system, is strikingly important in the diencephalon (thalamus and hypothalamus), the mesen- practice of genetic counseling. Early diagnosis is very cephalon forms the midbrain and rhombencephalon de- significant, not only because of the prognosis, but also velops into metencephalon (pons and cerebellum) and because of the emotional effects caused by the accompa- myelencephalon (medulla oblongata). At the time of the nying craniofacial malformations. The summary of the telencephalon/diencephalon differentiation, the prosen- obstetrical and diagnostical characteristics should be cephalon also splits longitudinally – the hemispheres de- useful in the management of holoprosencephaly. The velop on the lateral aspects of the longitudinal cerebral analysis of the 50 cases we encountered between 1981 fissure by progressive enlargement and hollowing of the and 2000, including the anatomical, diagnostic and clini- cerebral vesicles. Should the prechordal mesoderm fail to cal aspects, as well as the associated craniofacial malfor- migrate normally [1, 2], the prosencephalon remains un- mations, forms the essence of our publication. -

Version 1.0, 8/21/2016 Zika Pregnancy Outcome Reporting Of
Version 1.0, 8/21/2016 Zika Pregnancy outcome reporting of brain abnormalities and other adverse outcomes The following box details the inclusion criteria for brain abnormalities and other adverse outcomes potentially related to Zika virus infection during pregnancy. All pregnancy outcomes are monitored, but weekly reporting of adverse outcomes is limited to those meeting the below criteria. All prenatal and postnatal adverse outcomes are reported for both Zika Pregnancy Registries (US Zika Pregnancy Registry, Zika Active Pregnancy Surveillance System) and Active Birth Defects Surveillance; however, case finding methods dictate some differences in specific case definitions. Brain abnormalities with and without microcephaly Confirmed or possible congenital microcephaly# Intracranial calcifications Cerebral atrophy Abnormal cortical formation (e.g., polymicrogyria, lissencephaly, pachygyria, schizencephaly, gray matter heterotopia) Corpus callosum abnormalities Cerebellar abnormalities Porencephaly Hydranencephaly Ventriculomegaly / hydrocephaly (excluding “mild” ventriculomegaly without other brain abnormalities) Fetal brain disruption sequence (collapsed skull, overlapping sutures, prominent occipital bone, scalp rugae) Other major brain abnormalities, including intraventricular hemorrhage in utero (excluding post‐natal IVH) Early brain malformations, eye abnormalities, or consequences of central nervous system (CNS) dysfunction Neural tube defects (NTD) o Anencephaly / Acrania o Encephalocele o Spina bifida Holoprosencephaly -

Congenital Anomalies of the Nose
133 Congenital Anomalies of the Nose Jamie L. Funamura, MD1 Travis T. Tollefson, MD, MPH, FACS2 1 Department of Otolaryngology and Communication Enhancement, Address for correspondence Travis T. Tollefson, MD, MPH, FACS, Facial Children’s Hospital Boston, Boston, Massachusetts Plastic and Reconstructive Surgery, Department of Otolaryngology- 2 Department of Otolaryngology, University of California, Davis, Head and Neck Surgery, University of California, Davis, 2521 Stockton Sacramento, California Blvd., Suite 7200, Sacramento, CA 95817 (e-mail: [email protected]). Facial Plast Surg 2016;32:133–141. Abstract Congenital anomalies of the nose range from complete aplasia of the nose to duplications and nasal masses. Nasal development is the result of a complex embryo- logic patterning and fusion of multiple primordial structures. Loss of signaling proteins or failure of migration or proliferation can result in structural anomalies with significant Keywords cosmetic and functional consequences. Congenital anomalies of the nose can be ► nasal deformities categorized into four broad categories: (1) aplastic or hypoplastic, (2) hyperplastic or ► nasal dermoid duplications, (3) clefts, and (4) nasal masses. Our knowledge of the embryologic origin ► Tessier cleft of these anomalies helps dictate subsequent work-up for associated conditions, and the ► nasal cleft appropriate treatment or surgical approach to manage newborns and children with ► nasal hemangioma these anomalies. – Congenital anomalies of the nose are thought to be relatively side1 4 (►Fig. 1A, B). The medial processes will ultimately fuse, rare, affecting approximately 1 in every 20,000 to 40,000 live contributing to the nasal septum and the medial crura of the births.1 The exact incidence is difficult to quantify, as minor lower lateral cartilages. -
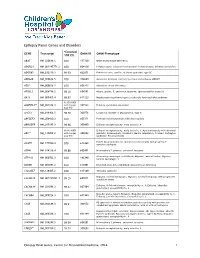
Epilepsy Panel Genes and Disorders
Epilepsy Panel Genes and Disorders *Covered GENE Transcript OMIM ID OMIM Phenotype 10X (%) ABAT NM_020686.5 100 137150 GABA-transaminase deficiency ADGRG1 NM_001145771.2 100 604110 Polymicrogyria, bilateral frontoparietal; Polymicrogyria, bilateral perisylvian ADGRV1 NM_032119.3 99.93 602851 Febrile seizures, familial, 4; Usher syndrome, type 2C ADRA2B NM_000682.5 100 104260 Autosomal dominant cortical myoclonus and epilepsy (ADCME) ADSL NM_000026.2 100 608222 Adenylosuccinase deficiency AFG3L2 NM_006796.2 98.16 604581 Ataxia, spastic, 5, autosomal recessive; Spinocerebellar ataxia 28 AKT3 NM_005465.4 99.97 611223 Megalencephaly-polymicrogyria-polydactyly-hydrocephalus syndrome 93.05 (100% ALDH7A1** NM_001182.3 with Sanger 107323 Epilepsy, pyridoxine-dependent Gap fill) ALG13 NM_018466.5 98.76 300776 Congenital disorder of glycosylation, type Is ARFGEF2 NM_006420.2 100 605371 Periventricular heterotopia with microcephaly ARHGEF9 NM_015185.3 99.96 300430 Epileptic encephalopathy, early infantile, 8 98.96 (100% Epileptic encephalopathy, early infantile, 1; Hydranencephaly with abnormal ARX** NM_139058.2 with Sanger 300382 genitalia; Lissencephaly, X-linked 2; Mental retardation, X-linked; Partington Gap fill) syndrome; Proud syndrome ASAH1 NM_177924.3 613468 Farber lipogranulomatosis; Spinal muscular atrophy with progressive 100 myoclonic epilepsy ASPM NM_018136.4 99.88 605481 Microcephaly 5, primary, autosomal recessive ATP1A2 NM_000702.3 182340 Alternating hemiplegia of childhood; Migraine, familial basilar; Migraine, 100 familial hemiplegic, -

Chapter III: Case Definition
NBDPN Guidelines for Conducting Birth Defects Surveillance rev. 06/04 Appendix 3.5 Case Inclusion Guidance for Potentially Zika-related Birth Defects Appendix 3.5 A3.5-1 Case Definition NBDPN Guidelines for Conducting Birth Defects Surveillance rev. 06/04 Appendix 3.5 Case Inclusion Guidance for Potentially Zika-related Birth Defects Contents Background ................................................................................................................................................. 1 Brain Abnormalities with and without Microcephaly ............................................................................. 2 Microcephaly ............................................................................................................................................................ 2 Intracranial Calcifications ......................................................................................................................................... 5 Cerebral / Cortical Atrophy ....................................................................................................................................... 7 Abnormal Cortical Gyral Patterns ............................................................................................................................. 9 Corpus Callosum Abnormalities ............................................................................................................................. 11 Cerebellar abnormalities ........................................................................................................................................ -

Blueprint Genetics Neuronal Migration Disorder Panel
Neuronal Migration Disorder Panel Test code: MA2601 Is a 59 gene panel that includes assessment of non-coding variants. Is ideal for patients with a clinical suspicion of neuronal migration disorder. About Neuronal Migration Disorder Neuronal migration disorders (NMDs) are a group of birth defects caused by the abnormal migration of neurons in the developing brain and nervous system. During development, neurons must migrate from the areas where they are originate to the areas where they will settle into their proper neural circuits. The structural abnormalities found in NMDs include schizencephaly, porencephaly, lissencephaly, agyria, macrogyria, polymicrogyria, pachygyria, microgyria, micropolygyria, neuronal heterotopias, agenesis of the corpus callosum, and agenesis of the cranial nerves. Mutations of many genes are involved in neuronal migration disorders, such as DCX in classical lissencephaly spectrum, TUBA1A in microlissencephaly with agenesis of the corpus callosum, and RELN and VLDLR in lissencephaly with cerebellar hypoplasia. Mutations in ARX cause a variety of phenotypes ranging from hydranencephaly or lissencephaly to early-onset epileptic encephalopathies, including Ohtahara syndrome and infantile spasms or intellectual disability with no brain malformations. Availability 4 weeks Gene Set Description Genes in the Neuronal Migration Disorder Panel and their clinical significance Gene Associated phenotypes Inheritance ClinVar HGMD ACTB* Baraitser-Winter syndrome AD 55 60 ACTG1* Deafness, Baraitser-Winter syndrome AD 27 47 ADGRG1 -

The Pathogenesis of Microcephaly Resulting from Congenital Infections: Why Is My Baby’S Head So Small?
Eur J Clin Microbiol Infect Dis DOI 10.1007/s10096-017-3111-8 REVIEW The pathogenesis of microcephaly resulting from congenital infections: why is my baby’s head so small? L. D. Frenkel1,2 & F. Gomez3 & F. Sabahi 4 Received: 29 August 2017 /Accepted: 17 September 2017 # Springer-Verlag GmbH Germany 2017 Abstract The emergence of Zika-virus-associated congenital review, we integrate all these findings to create a unified hy- microcephaly has engendered renewed interest in the patho- pothesis of the pathogenesis of congenital microcephaly in- genesis of microcephaly induced by infectious agents. Three duced by these infectious agents. of the original “TORCH” agents are associated with an appre- ciable incidence of congenital microcephaly: cytomegalovi- rus, rubella virus, and Toxoplasma gondii. The pathology of Introduction congenital microcephaly is characterized by neurotropic infec- tious agents that involve the fetal nervous system, leading to Microcephaly has become an issue of increased interest since brain destruction with calcifications, microcephaly, sensori- the recognition that it is a consequential manifestation of con- neural hearing loss, and ophthalmologic abnormalities. The genital Zika virus infection [1–3]. Microcephaly is generally inflammatory reaction induced by these four agents has an defined as a head circumference ≤ 2 standard deviations below important role in pathogenesis. The potential role of “strain the mean for gestational age [4]. Zika virus associated micro- differences” in pathogenesis of microcephaly by these four cephaly and other clinical manifestations of vertical transmis- pathogens is examined. Specific epidemiologic factors, such sion from mother to fetus during gestation are similar to those as first and early second trimester maternal infection, and the caused by three of the original “TORCH” agents (Toxoplasma manifestations of congenital infection in the infant, shed some gondii, rubella virus, and cytomegalovirus) [3, 5–7]. -

Hydranencephaly in Association with Roberts Syndrome
LE JOURNAL CANAD1EN DES SCIENCES NEUROLOGIQUKS Hydranencephaly in Association with Roberts Syndrome CHRIS E. U. EKONG AND BOHDAN ROZDILSKY SUMMARY: A clinicopathological In 1919, Dr. John Roberts de perforate. Marked hypertelorism study in a case of Roberts syndrome (tet- scribed a brother and sister with tet- was noted. The head appeared nor raphocomelia, cleft lip and palate, and raphocomelia, cleft lip and palate, mal in size but the neck was short and phallic hypertrophy) is reported. This pa and phallic hypertrophy. This com stiff. The infant died a few minutes tient had hydranencephaly and imperfo bination of congenital abnormalities after birth. Family history revealed rate anus, two additional congenital ab has subsequently been named that the patient's mother's two sis normalities so far not reported in this ters had no children, as they spon syndrome. Roberts syndrome. Twenty-two similar cases have been reported taneously aborted during each preg RESUME: Nous rapportons Vetude (Appelt et al., 1966; Holmes et al., nancy. The patient's parents were clinico pathologique d'un cas de syn 1972; Freeman et al., 1974). not consanguineously related. drome de Roberts (tetraphocomelie, bee Other congenital anomalies in A post mortem total body radio de lievre et hypertrophic phallique). Ce clude cryptorchidism, patent ductus graph (Figure 2) revealed ox patient presentait egalement une arteriosus and foramen ovale, en- ycephalic calvarium. The tubular hydranencephalie et tin anus non perfore cephalocele, polycystic kidneys, bones of the upper and lower ex — deux anomalies congenitales non horse-shoe kidneys and deformity of tremities were moderately well de prealablement connues dans ce syn base of skull and cribriform plate veloped, but were short compared to drome. -

Plastic Surgery Considerations for Holoprosencephaly Patients
Plastic Surgery Considerations for Holoprosencephaly Patients Jennifer M. Hendi, MPH* Robert Nemerofsky, MD* Cynthia Stolman, PhD† Mark S. Granick, MD* Newark, New Jersey Holoprosencephaly (HPE) is considered the lead- and upper lip, with normal or near-normal brain de- ing abnormality of the brain and face in humans velopment. Some data indicate that patients with less and is frequently associated with a wide spectrum severe manifestations of holoprosencephaly (i.e., of specific craniofacial anomalies including mid- semilobar and lobar) have survived into adulthood.3 line facial clefts, cyclopia and nasal irregularities. A A standard course of treatment of holoprosenceph- standard course of treatment has not been devel- aly has not been developed, and treatment is symp- oped and management is symptomatic and sup- tomatic and supportive. portive. In this work, the authors discuss the wide- Few guidelines have been described for surgical ranging spectrum of HPE and propose surgical candidates, resulting in conflicting recommendations guidelines to provide more uniform and appropri- as well as ethical and practical dilemmas for sur- ate care to patients suffering from holoprosenceph- geons. Given the prevalence and the wide-ranging manifestations of this disease, surgical guidelines aly. Assessment of the patient’s brain abnormality should be established to provide more uniform and is essential in determining the extent and benefit of appropriate care to patients suffering from holo- surgical intervention. The authors discuss a median prosencephaly. Assessment of the patient’s brain ab- straight-line repair of the lip and repair of the an- normality is essential in determining the extent and terior palate in a one-year old female and review benefit of surgical intervention.