The Expanding Phenotype of COL4A1 and COL4A2 Mutations: Clinical Data on 13 Newly Identified Families and a Review of the Literature
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Congenital Externally Communicating Porencephaly Presenting As Hemiplegic Cerebral Palsy: Imaging Study of a Rare Condition
SunKrist Journal of Neonatology and Pediatrics Case Presentation Volume: 3, Issue: 1 Scientific Knowledge Congenital Externally Communicating Porencephaly Presenting as Hemiplegic Cerebral Palsy: Imaging Study of a Rare Condition Al-Mosawi AJ1,2* 1Department of Pediatrics and Pediatric Psychiatry, Children Teaching Hospital of Baghdad Medical City, Iraq 2Head, Iraq Headquarter of Copernicus Scientists International Panel, Iraq 1. Abstract presentation including asymptomatic, various forms Congenital porencephaly is a very rare condition of cerebral palsy, seizures and cognitive impairment. characterized by cystic degeneration The disorder is heterogeneous in nature and the brain encephalomalacia and cysts or cavities within the lesions can be caused by developmental brain. Porencephalic cysts have a variable size and abnormalities, infection, perinatal brain ischemia, site and therefore it result in a variable clinical trauma and hemorrhage. Genetic factors have been presentations including asymptomatic, various forms suggested and familial cases have been reported. of cerebral palsy, seizures and cognitive impairment. Congenital porencephaly is generally classified into, The disorder is heterogeneous in nature and the brain internally communicating with the ventricle and lesions can be caused by developmental externally communicating with the subarachnoid abnormalities, infection, perinatal brain ischemia, space [1-7]. The aim of this paper is to report the rare trauma and hemorrhage. Genetic factors have been finding of externally communicating porencephaly in suggested and familial cases have been reported. a child with hemiplegic cerebral palsy. Congenital porencephaly is generally classified into, 4. Patients and Methods internally communicating with the ventricle and The case of a five-year old girl with hemiplegic externally communicating with the subarachnoid cerebral palsy caused by porencephaly is described space. -

Porencephaly Diagnosed by Isotope Cisternography
Journal of Neurology, Neurosurgery, and Psychiatry, 1972, 35, 669-675 J Neurol Neurosurg Psychiatry: first published as 10.1136/jnnp.35.5.669 on 1 October 1972. Downloaded from Porencephaly diagnosed by isotope cisternography D. FRONT, J. W. F. BEKS, AND L. PENNING' From the Departments of Neuroradiology and Neurosurgery, University Hospital, Gronintgen, The Netherlands SUMMARY The diagnosis of porencephaly by isotope cisternography is described. In the three cases presented, porencephaly was associated with non-resorptive hydrocephalus. The communi- cating hydrocephalus caused the isotope to enter the ventricular system and visualize the cyst, and the diagnosis of both disorders was established by RIHSA cisternography. The method is simple and non-traumatic and provides information about abnormalities which air may fail to demonstrate. Isotope cisternography has proved to be very plane of the collimator than does the gamma camera. useful in the diagnosis of disturbances of flow Every patient is studied at four, 24, and 48 hours and absorption of cerebrospinal fluid (CSF) and after injection. their resultant hydrocephalus (Di Chiro, Reames, and Matthews, 1964; Bannister, Gliford, and CASE 1 and Protected by copyright. Kocen, 1967; James, DeLand, Hodges, Wag- A 39 year old man suffered a head injury in a road ner, 1970; Front, 1971), and in the recognition of accident. Bleeding was noticed from his nose and CSF rhinorrhoea (Di Chiro and Grove, 1966; mouth but no abnormality was found on neurologi- Di Chiro, Ommaya, Ashburn, and Briner, 1968; cal examination. Plain radiographs of the skull Front and Penning, 1971). showed fractures of the nasal, right maxillary, and In addition, we have found this investigation zygomatic bones. -

CONGENITAL ABNORMALITIES of the CENTRAL NERVOUS SYSTEM Christopher Verity, Helen Firth, Charles Ffrench-Constant *I3
J Neurol Neurosurg Psychiatry: first published as 10.1136/jnnp.74.suppl_1.i3 on 1 March 2003. Downloaded from CONGENITAL ABNORMALITIES OF THE CENTRAL NERVOUS SYSTEM Christopher Verity, Helen Firth, Charles ffrench-Constant *i3 J Neurol Neurosurg Psychiatry 2003;74(Suppl I):i3–i8 dvances in genetics and molecular biology have led to a better understanding of the control of central nervous system (CNS) development. It is possible to classify CNS abnormalities Aaccording to the developmental stages at which they occur, as is shown below. The careful assessment of patients with these abnormalities is important in order to provide an accurate prog- nosis and genetic counselling. c NORMAL DEVELOPMENT OF THE CNS Before we review the various abnormalities that can affect the CNS, a brief overview of the normal development of the CNS is appropriate. c Induction—After development of the three cell layers of the early embryo (ectoderm, mesoderm, and endoderm), the underlying mesoderm (the “inducer”) sends signals to a region of the ecto- derm (the “induced tissue”), instructing it to develop into neural tissue. c Neural tube formation—The neural ectoderm folds to form a tube, which runs for most of the length of the embryo. c Regionalisation and specification—Specification of different regions and individual cells within the neural tube occurs in both the rostral/caudal and dorsal/ventral axis. The three basic regions of copyright. the CNS (forebrain, midbrain, and hindbrain) develop at the rostral end of the tube, with the spinal cord more caudally. Within the developing spinal cord specification of the different popu- lations of neural precursors (neural crest, sensory neurones, interneurones, glial cells, and motor neurones) is observed in progressively more ventral locations. -

Classification of Congenital Abnormalities of the CNS
315 Classification of Congenital Abnormalities of the CNS M. S. van der Knaap1 A classification of congenital cerebral, cerebellar, and spinal malformations is pre J . Valk2 sented with a view to its practical application in neuroradiology. The classification is based on the MR appearance of the morphologic abnormalities, arranged according to the embryologic time the derangement occurred. The normal embryology of the brain is briefly reviewed, and comments are made to explain the classification. MR images illustrating each subset of abnormalities are presented. During the last few years, MR imaging has proved to be a diagnostic tool of major importance in children with congenital malformations of the eNS [1]. The excellent gray fwhite-matter differentiation and multi planar imaging capabilities of MR allow a systematic analysis of the condition of the brain in infants and children. This is of interest for estimating prognosis and for genetic counseling. A classification is needed to serve as a guide to the great diversity of morphologic abnormalities and to make the acquired data useful. Such a system facilitates encoding, storage, and computer processing of data. We present a practical classification of congenital cerebral , cerebellar, and spinal malformations. Our classification is based on the morphologic abnormalities shown by MR and on the time at which the derangement of neural development occurred. A classification based on etiology is not as valuable because the various presumed causes rarely lead to a specific pattern of malformations. The abnor malities reflect the time the noxious agent interfered with neural development, rather than the nature of the noxious agent. The vulnerability of the various structures to adverse agents is greatest during the period of most active growth and development. -

Neonatal Porencephaly and Adult Stroke Related to Mutations in Collagen IV A1
Neonatal Porencephaly and Adult Stroke Related to Mutations in Collagen IV A1 Marjo S. van der Knaap, MD, PhD,1 Leo M. E. Smit, MD, PhD,1 Frederik Barkhof, MD, PhD,2 Yolande A. L. Pijnenburg, MD,3 Sonja Zweegman, MD, PhD,4 Hans W. M. Niessen, MD, PhD,5 Saskia Imhof, MD, PhD,6 and Peter Heutink, PhD7 Objective: The objective of this study was to describe leukoencephalopathy, lacunar infarcts, microbleeds and macro- bleeds in the context of a collagen IV A1 mutation. Methods: We examined a family with autosomal dominant poren- cephaly, in whom a defect in collagen IV A1 was detected recently. The patients underwent neurological, ophthalmo- logical, and cardiological examinations and magnetic resonance imaging of the brain. Electron microscopy of a skin biopsy was performed. Extensive laboratory screening was performed for thrombophilia and increased bleeding tendency. Results: The porencephaly was symptomatic in the infantile period in two patients, whereas it led to only minor neu- rological dysfunction in their affected mother. However, she experienced development of recurrent strokes in her 40s. In addition to the porencephaly, all patients had a leukoencephalopathy, which was most severe in the mother. Her mag- netic resonance imaging results also showed lacunar infarcts, macrobleeds and a multitude of microbleeds. No other risk factors for recurrent stroke were found. Electron microscopy showed interruptions of the basement membrane of skin capillaries and inhomogeneous thickening of the basement membrane with pools of basement membrane fragments. Interpretation: Leukoencephalopathy, ischemic infarcts, microbleeds, and macrobleeds are indicative of an underlying microangiopathy, of which the best-known causes are hypertension, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy, and cerebral amyloid angiopathy. -

Lack of Serologic Evidence for an Association Between Cache Valley Virus Infection and Anencephaly and Other Neural Tube Defects in Texas
Dispatches Lack of Serologic Evidence for an Association between Cache Valley Virus Infection and Anencephaly and other Neural Tube Defects in Texas We tested the hypothesis that Cache Valley Virus (CVV), an endemic North American bunyavirus, may be involved in the pathogenesis of human neural tube defects. This investigation followed a 1990 and 1991 south Texas outbreak of neural tube defects with a high prevalence of anencephaly and the demonstration in 1987 that in utero infection by CVV was the cause of outbreaks of central nervous system and musculoskeletal defects in North American ruminants. Sera from 74 women who gave birth to infants with neural tube defects in south Texas from 1993 through early 1995 were tested for CVV neutralizing antibody. All tested sera did not neutralize CVV. These data suggest that CVV is not involved in the induction of human neural tube defects during nonepidemic periods but do not preclude CVV involvement during epidemics. Other endemic bunyaviruses may still be involved in the pathogenesis of neural tube defects or other congenital central nervous system or musculoskeletal malformations. Anencephaly, spina bifida, and encephalocele showed that in south Texas during this period the (the major types of neural tube defects) are average annual prevalence of anencephaly was generally due to the failure of the neural tube to approximately 4.9 per 10,000 births. Women with close during early embryonic development (1). Hispanic surnames, three or more previous live Neural tube defects are among the most common births, history of stillbirth, or residence in east or and most severe major birth defects. -

Version 1.0, 8/21/2016 Zika Pregnancy Outcome Reporting Of
Version 1.0, 8/21/2016 Zika Pregnancy outcome reporting of brain abnormalities and other adverse outcomes The following box details the inclusion criteria for brain abnormalities and other adverse outcomes potentially related to Zika virus infection during pregnancy. All pregnancy outcomes are monitored, but weekly reporting of adverse outcomes is limited to those meeting the below criteria. All prenatal and postnatal adverse outcomes are reported for both Zika Pregnancy Registries (US Zika Pregnancy Registry, Zika Active Pregnancy Surveillance System) and Active Birth Defects Surveillance; however, case finding methods dictate some differences in specific case definitions. Brain abnormalities with and without microcephaly Confirmed or possible congenital microcephaly# Intracranial calcifications Cerebral atrophy Abnormal cortical formation (e.g., polymicrogyria, lissencephaly, pachygyria, schizencephaly, gray matter heterotopia) Corpus callosum abnormalities Cerebellar abnormalities Porencephaly Hydranencephaly Ventriculomegaly / hydrocephaly (excluding “mild” ventriculomegaly without other brain abnormalities) Fetal brain disruption sequence (collapsed skull, overlapping sutures, prominent occipital bone, scalp rugae) Other major brain abnormalities, including intraventricular hemorrhage in utero (excluding post‐natal IVH) Early brain malformations, eye abnormalities, or consequences of central nervous system (CNS) dysfunction Neural tube defects (NTD) o Anencephaly / Acrania o Encephalocele o Spina bifida Holoprosencephaly -
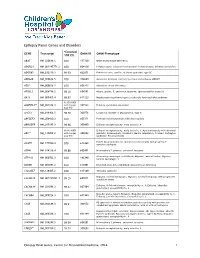
Epilepsy Panel Genes and Disorders
Epilepsy Panel Genes and Disorders *Covered GENE Transcript OMIM ID OMIM Phenotype 10X (%) ABAT NM_020686.5 100 137150 GABA-transaminase deficiency ADGRG1 NM_001145771.2 100 604110 Polymicrogyria, bilateral frontoparietal; Polymicrogyria, bilateral perisylvian ADGRV1 NM_032119.3 99.93 602851 Febrile seizures, familial, 4; Usher syndrome, type 2C ADRA2B NM_000682.5 100 104260 Autosomal dominant cortical myoclonus and epilepsy (ADCME) ADSL NM_000026.2 100 608222 Adenylosuccinase deficiency AFG3L2 NM_006796.2 98.16 604581 Ataxia, spastic, 5, autosomal recessive; Spinocerebellar ataxia 28 AKT3 NM_005465.4 99.97 611223 Megalencephaly-polymicrogyria-polydactyly-hydrocephalus syndrome 93.05 (100% ALDH7A1** NM_001182.3 with Sanger 107323 Epilepsy, pyridoxine-dependent Gap fill) ALG13 NM_018466.5 98.76 300776 Congenital disorder of glycosylation, type Is ARFGEF2 NM_006420.2 100 605371 Periventricular heterotopia with microcephaly ARHGEF9 NM_015185.3 99.96 300430 Epileptic encephalopathy, early infantile, 8 98.96 (100% Epileptic encephalopathy, early infantile, 1; Hydranencephaly with abnormal ARX** NM_139058.2 with Sanger 300382 genitalia; Lissencephaly, X-linked 2; Mental retardation, X-linked; Partington Gap fill) syndrome; Proud syndrome ASAH1 NM_177924.3 613468 Farber lipogranulomatosis; Spinal muscular atrophy with progressive 100 myoclonic epilepsy ASPM NM_018136.4 99.88 605481 Microcephaly 5, primary, autosomal recessive ATP1A2 NM_000702.3 182340 Alternating hemiplegia of childhood; Migraine, familial basilar; Migraine, 100 familial hemiplegic, -

Chapter III: Case Definition
NBDPN Guidelines for Conducting Birth Defects Surveillance rev. 06/04 Appendix 3.5 Case Inclusion Guidance for Potentially Zika-related Birth Defects Appendix 3.5 A3.5-1 Case Definition NBDPN Guidelines for Conducting Birth Defects Surveillance rev. 06/04 Appendix 3.5 Case Inclusion Guidance for Potentially Zika-related Birth Defects Contents Background ................................................................................................................................................. 1 Brain Abnormalities with and without Microcephaly ............................................................................. 2 Microcephaly ............................................................................................................................................................ 2 Intracranial Calcifications ......................................................................................................................................... 5 Cerebral / Cortical Atrophy ....................................................................................................................................... 7 Abnormal Cortical Gyral Patterns ............................................................................................................................. 9 Corpus Callosum Abnormalities ............................................................................................................................. 11 Cerebellar abnormalities ........................................................................................................................................ -

Blueprint Genetics Neuronal Migration Disorder Panel
Neuronal Migration Disorder Panel Test code: MA2601 Is a 59 gene panel that includes assessment of non-coding variants. Is ideal for patients with a clinical suspicion of neuronal migration disorder. About Neuronal Migration Disorder Neuronal migration disorders (NMDs) are a group of birth defects caused by the abnormal migration of neurons in the developing brain and nervous system. During development, neurons must migrate from the areas where they are originate to the areas where they will settle into their proper neural circuits. The structural abnormalities found in NMDs include schizencephaly, porencephaly, lissencephaly, agyria, macrogyria, polymicrogyria, pachygyria, microgyria, micropolygyria, neuronal heterotopias, agenesis of the corpus callosum, and agenesis of the cranial nerves. Mutations of many genes are involved in neuronal migration disorders, such as DCX in classical lissencephaly spectrum, TUBA1A in microlissencephaly with agenesis of the corpus callosum, and RELN and VLDLR in lissencephaly with cerebellar hypoplasia. Mutations in ARX cause a variety of phenotypes ranging from hydranencephaly or lissencephaly to early-onset epileptic encephalopathies, including Ohtahara syndrome and infantile spasms or intellectual disability with no brain malformations. Availability 4 weeks Gene Set Description Genes in the Neuronal Migration Disorder Panel and their clinical significance Gene Associated phenotypes Inheritance ClinVar HGMD ACTB* Baraitser-Winter syndrome AD 55 60 ACTG1* Deafness, Baraitser-Winter syndrome AD 27 47 ADGRG1 -

Syndromes, Disorders and Maternal Risk Factors Associated with Neural Tube Defects (Vii)
■ REVIEW ARTICLE ■ SYNDROMES, DISORDERS AND MATERNAL RISK FACTORS ASSOCIATED WITH NEURAL TUBE DEFECTS (VII) Chih-Ping Chen1,2,3,4,5* 1Department of Obstetrics and Gynecology, and 2Medical Research, Mackay Memorial Hospital, Taipei, 3Department of Biotechnology, Asia University, 4School of Chinese Medicine, College of Chinese Medicine, China Medical University, Taichung, and 5Institute of Clinical and Community Health Nursing, National Yang-Ming University, Taipei, Taiwan. SUMMARY Neural tube defects (NTDs) may be associated with syndromes, disorders and maternal risk factors. This article provides a comprehensive review of the syndromes, disorders and maternal risk factors associated with NTDs, including DK phocomelia syndrome (von Voss-Cherstvoy syndrome), Siegel-Bartlet syndrome, fetal warfarin syndrome, craniotelencephalic dysplasia, Czeizel-Losonci syndrome, maternal cocaine abuse, Weissenbacher- Zweymüller syndrome, parietal foramina (cranium bifidum), Apert syndrome, craniomicromelic syndrome, XX- agonadism with multiple dysraphic lesions including omphalocele and NTDs, Fryns microphthalmia syndrome, Gershoni-Baruch syndrome, PHAVER syndrome, periconceptional vitamin B6 deficiency, and autosomal dominant Dandy-Walker malformation with occipital cephalocele. NTDs associated with these syndromes, disorders and maternal risk factors are a rare but important cause of NTDs. The recurrence risk and the preventive effect of mater- nal folic acid intake in NTDs associated with syndromes, disorders and maternal risk factors may be different -

Hydranencephaly
Kathmandu University Medical Journal (2010), Vol. 8, No. 1, Issue 29, 83-86 Case Note Hydranencephaly Pant S1, Kaur G2, JK De3 1Medical Offi cer, 2Associate Professor, 3Professor and Head, Department of Obstetrics and Gynaecology, Manipal College of Medical Sciences Abstract Hydranencephaly is a rare congenital condition where the greater portions of the cerebral hemispheres and the corpus striatum are replaced by cerebrospinal fl uid and glial tissue. The meninges and the skull are well formed, which is consistent with earlier normal embryogenesis of the telencephalon. Bilateral occlusion of the internal carotid arteries in utero is a potential mechanism. Clinical features include intact brainstem refl exes without evidence of higher cortical activity. The infant’s head size and the spontaneous refl exes such as sucking, swallowing, crying, and moving the arms and legs may all seem normal at birth. However, after a few weeks the infant usually becomes irritable and has increased muscle tone and after a few months of life, seizures and hydrocephalus (excessive accumulation of cerebrospinal fl uid in the brain) may develop. Other symptoms may include visual impairment, lack of growth, deafness, blindness, spastic quadriparesis (paralysis), and intellectual defi cits. Since the early behaviour appears to be relatively normal, the diagnosis may be delayed for months sometimes. There is no defi nitive treatment for hydranencephaly. The outlook for children with hydranencephaly is generally poor, and many children with this disorder die before their fi rst birthday. Key words: hydranencephaly, congenital anomaly, vascular disruption, thromboplastin, 19 years old, Gravida 2 Para 0 Abortion 1, presented HBV, blood sugars) were normal.