207768Orig1s000
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

(CD-P-PH/PHO) Report Classification/Justifica
COMMITTEE OF EXPERTS ON THE CLASSIFICATION OF MEDICINES AS REGARDS THEIR SUPPLY (CD-P-PH/PHO) Report classification/justification of medicines belonging to the ATC group R01 (Nasal preparations) Table of Contents Page INTRODUCTION 5 DISCLAIMER 7 GLOSSARY OF TERMS USED IN THIS DOCUMENT 8 ACTIVE SUBSTANCES Cyclopentamine (ATC: R01AA02) 10 Ephedrine (ATC: R01AA03) 11 Phenylephrine (ATC: R01AA04) 14 Oxymetazoline (ATC: R01AA05) 16 Tetryzoline (ATC: R01AA06) 19 Xylometazoline (ATC: R01AA07) 20 Naphazoline (ATC: R01AA08) 23 Tramazoline (ATC: R01AA09) 26 Metizoline (ATC: R01AA10) 29 Tuaminoheptane (ATC: R01AA11) 30 Fenoxazoline (ATC: R01AA12) 31 Tymazoline (ATC: R01AA13) 32 Epinephrine (ATC: R01AA14) 33 Indanazoline (ATC: R01AA15) 34 Phenylephrine (ATC: R01AB01) 35 Naphazoline (ATC: R01AB02) 37 Tetryzoline (ATC: R01AB03) 39 Ephedrine (ATC: R01AB05) 40 Xylometazoline (ATC: R01AB06) 41 Oxymetazoline (ATC: R01AB07) 45 Tuaminoheptane (ATC: R01AB08) 46 Cromoglicic Acid (ATC: R01AC01) 49 2 Levocabastine (ATC: R01AC02) 51 Azelastine (ATC: R01AC03) 53 Antazoline (ATC: R01AC04) 56 Spaglumic Acid (ATC: R01AC05) 57 Thonzylamine (ATC: R01AC06) 58 Nedocromil (ATC: R01AC07) 59 Olopatadine (ATC: R01AC08) 60 Cromoglicic Acid, Combinations (ATC: R01AC51) 61 Beclometasone (ATC: R01AD01) 62 Prednisolone (ATC: R01AD02) 66 Dexamethasone (ATC: R01AD03) 67 Flunisolide (ATC: R01AD04) 68 Budesonide (ATC: R01AD05) 69 Betamethasone (ATC: R01AD06) 72 Tixocortol (ATC: R01AD07) 73 Fluticasone (ATC: R01AD08) 74 Mometasone (ATC: R01AD09) 78 Triamcinolone (ATC: R01AD11) 82 -

Prohibited Substances List
Prohibited Substances List This is the Equine Prohibited Substances List that was voted in at the FEI General Assembly in November 2009 alongside the new Equine Anti-Doping and Controlled Medication Regulations(EADCMR). Neither the List nor the EADCM Regulations are in current usage. Both come into effect on 1 January 2010. The current list of FEI prohibited substances remains in effect until 31 December 2009 and can be found at Annex II Vet Regs (11th edition) Changes in this List : Shaded row means that either removed or allowed at certain limits only SUBSTANCE ACTIVITY Banned Substances 1 Acebutolol Beta blocker 2 Acefylline Bronchodilator 3 Acemetacin NSAID 4 Acenocoumarol Anticoagulant 5 Acetanilid Analgesic/anti-pyretic 6 Acetohexamide Pancreatic stimulant 7 Acetominophen (Paracetamol) Analgesic/anti-pyretic 8 Acetophenazine Antipsychotic 9 Acetylmorphine Narcotic 10 Adinazolam Anxiolytic 11 Adiphenine Anti-spasmodic 12 Adrafinil Stimulant 13 Adrenaline Stimulant 14 Adrenochrome Haemostatic 15 Alclofenac NSAID 16 Alcuronium Muscle relaxant 17 Aldosterone Hormone 18 Alfentanil Narcotic 19 Allopurinol Xanthine oxidase inhibitor (anti-hyperuricaemia) 20 Almotriptan 5 HT agonist (anti-migraine) 21 Alphadolone acetate Neurosteriod 22 Alphaprodine Opiod analgesic 23 Alpidem Anxiolytic 24 Alprazolam Anxiolytic 25 Alprenolol Beta blocker 26 Althesin IV anaesthetic 27 Althiazide Diuretic 28 Altrenogest (in males and gelidngs) Oestrus suppression 29 Alverine Antispasmodic 30 Amantadine Dopaminergic 31 Ambenonium Cholinesterase inhibition 32 Ambucetamide Antispasmodic 33 Amethocaine Local anaesthetic 34 Amfepramone Stimulant 35 Amfetaminil Stimulant 36 Amidephrine Vasoconstrictor 37 Amiloride Diuretic 1 Prohibited Substances List This is the Equine Prohibited Substances List that was voted in at the FEI General Assembly in November 2009 alongside the new Equine Anti-Doping and Controlled Medication Regulations(EADCMR). -

(19) United States (12) Patent Application Publication (10) Pub
US 20130210835A1 (19) United States (12) Patent Application Publication (10) Pub. N0.2 US 2013/0210835 A1 Mitchell (43) Pub. Date: Aug. 15, 2013 (54) PHARMACEUTICAL COMPOSITIONS Publication Classi?cation (75) Inventor: Odes W. Mitchell; Arlington, TX (U S) (51) Int. Cl. A61K31/137 (2006.01) _ A611; 31/4402 (2006.01) (73) Ass1gnee: GM PHARMACEUTICAL, INC, A61K 31/485 (200601) Arhngton, TX (Us) A611; 31/09 (2006.01) _ A611; 31/495 (2006.01) (21) App1.No.. 13/703,584 A61K31/505 (200601) 22 PCT P1 d: J .13 2011 (52) us Cl ( ) 1e “n ’ CPC ........... .. A611; 31/137 (2013.01); A611;31/495 (86) PCT NO. PCT/“11,4031 (2013.01); A611;31/505 (2013.01); A611; 31/485 (2013.01); A611; 31/09 (2013.01); § 371 (0)0). A611;31/4402 (2013.01) (2), (4) Date: Feb- 2, 2013 USPC .... .. 514/255.04; 564/355; 514/653; 544/396; 544/332; 514/275; 546/74; 514/289; 514/282; Related US. Application Data 514657; 514652 (60) Provisional application No. 61/354,061; ?led on Jun. (57) ABSTRACT 11; 2010; provisional application No. 61/354,057; A composition of an antitussive; a decongestant; or an anti ?led on Jun. 11; 2010; provisional application No. histamine to treat respiratory and oral pharyngeal congestion 61/354,053; ?led on Jun. 11,2010. and related symptoms in a patient. US 2013/0210835 A1 Aug. 15,2013 PHARMACEUTICAL COMPOSITIONS mucus build-up to clear congestion in the air passages. Symp toms due to allergies or allergens are often treated With an CROSS-REFERENCES TO RELATED antihistamine. -

236 Subpart A—General Provisions Subpart B—Active Ingredients
§ 341.1 21 CFR Ch. I (4–1–01 Edition) 341.78 Labeling of expectorant drug prod- (g) Topical nasal decongestant drug. A ucts. drug that when applied topically inside 341.80 Labeling of nasal decongestant drug the nose, in the form of drops, jellies, products. or sprays, or when inhaled intranasally 341.90 Professional labeling. reduces nasal congestion caused by AUTHORITY: 21 U.S.C. 321, 351, 352, 353, 355, acute or chronic rhinitis. 360, 371. (h) Calibrated dropper. A dropper cali- brated such that the volume error in- Subpart A—General Provisions curred in measuring any liquid does not exceed 15 percent under normal use § 341.1 Scope. conditions. (a) An over-the-counter cold, cough, [51 FR 35339, Oct. 2, 1986, as amended at 54 FR allergy, bronchodilator, or anti- 8509, Feb. 28, 1989; 55 FR 40382, Oct. 3, 1990; 57 asthmatic drug product in a form suit- FR 58374, Dec. 9, 1992; 59 FR 43409, Aug. 23, able for oral, inhalant, or topical ad- 1994] ministration is generally recognized as safe and effective and is not mis- Subpart B—Active Ingredients branded if it meets each of the condi- tions in this part and each of the gen- § 341.12 Antihistamine active ingredi- eral conditions established in § 330.1. ents. (b) References in this part to regu- The active ingredient of the product latory sections of the Code of Federal consists of any of the following when Regulations are to chapter I of title 21 used within the dosage limits estab- unless otherwise noted. lished for each ingredient: [51 FR 35339, Oct. -
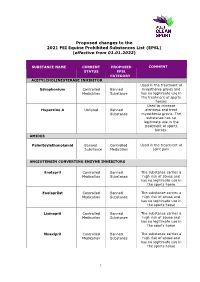
Proposed Changes to the 2021 FEI Equine Prohibited Substances List (EPSL) (Effective from 01.01.2022)
Proposed changes to the 2021 FEI Equine Prohibited Substances List (EPSL) (effective from 01.01.2022) SUBSTANCE NAME CURRENT PROPOSED COMMENT STATUS EPSL CATEGORY ACETYLCHOLINESTERASE INHIBITOR Used in the treatment of Edrophonium Controlled Banned myasthenia gravis and Medication Substance has no legitimate use in the treatment of sports horses Used to increase Huperzine A Unlisted Banned alertness and treat Substance myasthenia gravis. The substance has no legitimate use in the treatment of sports horses. AMIDES Palmitoylethanolamid Banned Controlled Used in the treatment of Substance Medication joint pain ANGIOTENSIN CONVERTING ENZYME INHIBITORS Enalapril Controlled Banned The substance carries a Medication Substance high risk of abuse and has no legitimate use in the sports horse Enalaprilat Controlled Banned The substance carries a Medication Substance high risk of abuse and has no legitimate use in the sports horse Lisinopril Controlled Banned The substance carries a Medication Substance high risk of abuse and has no legitimate use in the sports horse Moexipril Controlled Banned The substance carries a Medication Substance high risk of abuse and has no legitimate use in the sports horse 1 Perindoprilat Controlled Banned The substance carries a Medication Substance high risk of abuse and has no legitimate use in the sports horse ANTIHISTAMINES Antazoline Controlled Banned The substance has no Medication Substance legitimate use in the sports horse Azatadine Controlled Banned The substance has Medication Substance sedative effects -

BMJ Open Is Committed to Open Peer Review. As Part of This Commitment We Make the Peer Review History of Every Article We Publish Publicly Available
BMJ Open is committed to open peer review. As part of this commitment we make the peer review history of every article we publish publicly available. When an article is published we post the peer reviewers’ comments and the authors’ responses online. We also post the versions of the paper that were used during peer review. These are the versions that the peer review comments apply to. The versions of the paper that follow are the versions that were submitted during the peer review process. They are not the versions of record or the final published versions. They should not be cited or distributed as the published version of this manuscript. BMJ Open is an open access journal and the full, final, typeset and author-corrected version of record of the manuscript is available on our site with no access controls, subscription charges or pay-per-view fees (http://bmjopen.bmj.com). If you have any questions on BMJ Open’s open peer review process please email [email protected] BMJ Open Pediatric drug utilization in the Western Pacific region: Australia, Japan, South Korea, Hong Kong and Taiwan Journal: BMJ Open ManuscriptFor ID peerbmjopen-2019-032426 review only Article Type: Research Date Submitted by the 27-Jun-2019 Author: Complete List of Authors: Brauer, Ruth; University College London, Research Department of Practice and Policy, School of Pharmacy Wong, Ian; University College London, Research Department of Practice and Policy, School of Pharmacy; University of Hong Kong, Centre for Safe Medication Practice and Research, Department -

Vr Meds Ex01 3B 0825S Coding Manual Supplement Page 1
vr_meds_ex01_3b_0825s Coding Manual Supplement MEDNAME OTHER_CODE ATC_CODE SYSTEM THER_GP PHRM_GP CHEM_GP SODIUM FLUORIDE A12CD01 A01AA01 A A01 A01A A01AA SODIUM MONOFLUOROPHOSPHATE A12CD02 A01AA02 A A01 A01A A01AA HYDROGEN PEROXIDE D08AX01 A01AB02 A A01 A01A A01AB HYDROGEN PEROXIDE S02AA06 A01AB02 A A01 A01A A01AB CHLORHEXIDINE B05CA02 A01AB03 A A01 A01A A01AB CHLORHEXIDINE D08AC02 A01AB03 A A01 A01A A01AB CHLORHEXIDINE D09AA12 A01AB03 A A01 A01A A01AB CHLORHEXIDINE R02AA05 A01AB03 A A01 A01A A01AB CHLORHEXIDINE S01AX09 A01AB03 A A01 A01A A01AB CHLORHEXIDINE S02AA09 A01AB03 A A01 A01A A01AB CHLORHEXIDINE S03AA04 A01AB03 A A01 A01A A01AB AMPHOTERICIN B A07AA07 A01AB04 A A01 A01A A01AB AMPHOTERICIN B G01AA03 A01AB04 A A01 A01A A01AB AMPHOTERICIN B J02AA01 A01AB04 A A01 A01A A01AB POLYNOXYLIN D01AE05 A01AB05 A A01 A01A A01AB OXYQUINOLINE D08AH03 A01AB07 A A01 A01A A01AB OXYQUINOLINE G01AC30 A01AB07 A A01 A01A A01AB OXYQUINOLINE R02AA14 A01AB07 A A01 A01A A01AB NEOMYCIN A07AA01 A01AB08 A A01 A01A A01AB NEOMYCIN B05CA09 A01AB08 A A01 A01A A01AB NEOMYCIN D06AX04 A01AB08 A A01 A01A A01AB NEOMYCIN J01GB05 A01AB08 A A01 A01A A01AB NEOMYCIN R02AB01 A01AB08 A A01 A01A A01AB NEOMYCIN S01AA03 A01AB08 A A01 A01A A01AB NEOMYCIN S02AA07 A01AB08 A A01 A01A A01AB NEOMYCIN S03AA01 A01AB08 A A01 A01A A01AB MICONAZOLE A07AC01 A01AB09 A A01 A01A A01AB MICONAZOLE D01AC02 A01AB09 A A01 A01A A01AB MICONAZOLE G01AF04 A01AB09 A A01 A01A A01AB MICONAZOLE J02AB01 A01AB09 A A01 A01A A01AB MICONAZOLE S02AA13 A01AB09 A A01 A01A A01AB NATAMYCIN A07AA03 A01AB10 A A01 -

Federal Register / Vol. 60, No. 80 / Wednesday, April 26, 1995 / Notices DIX to the HTSUS—Continued
20558 Federal Register / Vol. 60, No. 80 / Wednesday, April 26, 1995 / Notices DEPARMENT OF THE TREASURY Services, U.S. Customs Service, 1301 TABLE 1.ÐPHARMACEUTICAL APPEN- Constitution Avenue NW, Washington, DIX TO THE HTSUSÐContinued Customs Service D.C. 20229 at (202) 927±1060. CAS No. Pharmaceutical [T.D. 95±33] Dated: April 14, 1995. 52±78±8 ..................... NORETHANDROLONE. A. W. Tennant, 52±86±8 ..................... HALOPERIDOL. Pharmaceutical Tables 1 and 3 of the Director, Office of Laboratories and Scientific 52±88±0 ..................... ATROPINE METHONITRATE. HTSUS 52±90±4 ..................... CYSTEINE. Services. 53±03±2 ..................... PREDNISONE. 53±06±5 ..................... CORTISONE. AGENCY: Customs Service, Department TABLE 1.ÐPHARMACEUTICAL 53±10±1 ..................... HYDROXYDIONE SODIUM SUCCI- of the Treasury. NATE. APPENDIX TO THE HTSUS 53±16±7 ..................... ESTRONE. ACTION: Listing of the products found in 53±18±9 ..................... BIETASERPINE. Table 1 and Table 3 of the CAS No. Pharmaceutical 53±19±0 ..................... MITOTANE. 53±31±6 ..................... MEDIBAZINE. Pharmaceutical Appendix to the N/A ............................. ACTAGARDIN. 53±33±8 ..................... PARAMETHASONE. Harmonized Tariff Schedule of the N/A ............................. ARDACIN. 53±34±9 ..................... FLUPREDNISOLONE. N/A ............................. BICIROMAB. 53±39±4 ..................... OXANDROLONE. United States of America in Chemical N/A ............................. CELUCLORAL. 53±43±0 -

Nasal Decongestant Active Ingredients
Food and Drug Administration, HHS § 341.20 (d) Expectorant drug. A drug taken (m) Triprolidine hydrochloride. orally to promote or facilitate the re- [57 FR 58374, Dec. 9, 1992, as amended at 59 moval of secretions from the res- FR 4218, Jan. 28, 1994] piratory airways. (e) Antihistamine drug. A drug used § 341.14 Antitussive active ingredients. for the relief of the symptoms of hay The active ingredients of the product fever and upper respiratory allergies consist of any of the following when (allergic rhinitis). used within the dosage limits and in (f) Oral nasal decongestant drug. A the dosage forms established for each drug that is taken by mouth and acts ingredient in § 341.74(d): systemically to reduce nasal conges- (a) Oral antitussives. (1) Chlophedianol tion caused by acute or chronic rhi- hydrochloride. nitis. (2) Codeine ingredients. The following (g) Topical nasal decongestant drug. A ingredients may be used only in com- drug that when applied topically inside bination in accordance with §§ 290.2 and the nose, in the form of drops, jellies, 21 CFR 1308.15(c). or sprays, or when inhaled intranasally (i) Codeine. reduces nasal congestion caused by (ii) Codeine phosphate. acute or chronic rhinitis. (iii) Codeine sulfate. (h) Calibrated dropper. A dropper cali- (3) Dextromethorphan. brated such that the volume error in- (4) Dextromethorphan hydrobromide. curred in measuring any liquid does (5) Diphenhydramine citrate. not exceed 15 percent under normal use (6) Diphenhydramine hydrochloride. conditions. (b) Topical antitussives. (1) Camphor. (i) Effervescent dosage form. A dosage (2) Menthol. form intended to be dissolved in water [52 FR 30055, Aug. -

209089Orig1s000 209090Orig1s000
CENTER FOR DRUG EVALUATION AND RESEARCH APPLICATION NUMBER: 209089Orig1s000 209090Orig1s000 CLINICAL REVIEW(S) CLINICAL REVIEW Application Type NDA (Rx to OTC switch) Application Number(s) 209-089 Xyzal Allergy 24 HR tablets 209-090 Xyzal Allergy 24 HR solution Priority or Standard Standard Submit Date(s) March 18, 2016 Received Date(s) March 31, 2016 PDUFA Goal Date January 31, 2017 Division / Office DPARP/ODE 2/OND Reviewer Name(s) Xu Wang, M.D., Ph.D. Review Completion Date December 9, 2016 Established Name Levocetirizine dihydrochloride Rx Trade Name Xyzal tablets/solution OTC Trade Name Xyzal Allergy 24HR tablets/solution Therapeutic Class Antihistamine Applicant UCB Inc. Formulation(s) tablets/solution Dosing Regimen ≥12 years: One tablet (5 mg) daily/10 mL solution (5 mg) daily; 1 6 – 11 years: /2 tablet (2.5 mg) daily/5 mL solution (2.5 mg) daily; 2 – 5 years: 2.5 mL solution (1.25 mg) daily Indication(s) Temporarily relieve these symptoms due to hay fever or other upper respiratory allergies: • running nose, • sneezing, • itchy, watering eyes, • itching of nose or throat Intended Population(s) Tablets: ≥6 years of age Solution: ≥2 years of age Template Version: March 6, 2009 Reference ID: 4025819 Clinical Review Xu Wang, M.D., Ph.D. NDA 209-089/NDA 209-090 Rx to OTC switch Xyzal Allergy 24HR (levocetirizine dihydrochloride) tablets/solution Table of Contents 1 RECOMMENDATIONS/RISK BENEFIT ASSESSMENT .........................................4 1.1 Recommendation on Regulatory Action ............................................................. 4 1.2 Risk Benefit Assessment .................................................................................... 4 2 INTRODUCTION AND REGULATORY BACKGROUND ........................................ 4 2.1 Product Information ............................................................................................ 4 2.2 Currently Available Treatments for Proposed Indications.................................. -

Prescription Medications, Drugs, Herbs & Chemicals Associated With
Prescription Medications, Drugs, Herbs & Chemicals Associated with Tinnitus American Tinnitus Association Prescription Medications, Drugs, Herbs & Chemicals Associated with Tinnitus All rights reserved. No part of this publication may be reproduced, stored in a retrieval system or transmitted in any form, or by any means, without the prior written permission of the American Tinnitus Association. ©2013 American Tinnitus Association Prescription Medications, Drugs, Herbs & Chemicals Associated with Tinnitus American Tinnitus Association This document is to be utilized as a conversation tool with your health care provider and is by no means a “complete” listing. Anyone reading this list of ototoxic drugs is strongly advised NOT to discontinue taking any prescribed medication without first contacting the prescribing physician. Just because a drug is listed does not mean that you will automatically get tinnitus, or exacerbate exisiting tinnitus, if you take it. A few will, but many will not. Whether or not you eperience tinnitus after taking one of the listed drugs or herbals, or after being exposed to one of the listed chemicals, depends on many factors ‐ such as your own body chemistry, your sensitivity to drugs, the dose you take, or the length of time you take the drug. It is important to note that there may be drugs NOT listed here that could still cause tinnitus. Although this list is one of the most complete listings of drugs associated with tinnitus, no list of this kind can ever be totally complete – therefore use it as a guide and resource, but do not take it as the final word. The drug brand name is italicized and is followed by the generic drug name in bold. -

Common Study Protocol for Observational Database Studies WP5 – Analytic Database Studies
Arrhythmogenic potential of drugs FP7-HEALTH-241679 http://www.aritmo-project.org/ Common Study Protocol for Observational Database Studies WP5 – Analytic Database Studies V 1.3 Draft Lead beneficiary: EMC Date: 03/01/2010 Nature: Report Dissemination level: D5.2 Report on Common Study Protocol for Observational Database Studies WP5: Conduct of Additional Observational Security: Studies. Author(s): Gianluca Trifiro’ (EMC), Giampiero Version: v1.1– 2/85 Mazzaglia (F-SIMG) Draft TABLE OF CONTENTS DOCUMENT INFOOMATION AND HISTORY ...........................................................................4 DEFINITIONS .................................................... ERRORE. IL SEGNALIBRO NON È DEFINITO. ABBREVIATIONS ......................................................................................................................6 1. BACKGROUND .................................................................................................................7 2. STUDY OBJECTIVES................................ ERRORE. IL SEGNALIBRO NON È DEFINITO. 3. METHODS ..........................................................................................................................8 3.1.STUDY DESIGN ....................................................................................................................8 3.2.DATA SOURCES ..................................................................................................................9 3.2.1. IPCI Database .....................................................................................................9