Don't Worry, Be Happy: Endocannabinoids and Cannabis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Cannabinoids and Endocannabinoid System Changes in Intestinal Inflammation and Colorectal Cancer
cancers Review Cannabinoids and Endocannabinoid System Changes in Intestinal Inflammation and Colorectal Cancer Viktoriia Cherkasova, Olga Kovalchuk * and Igor Kovalchuk * Department of Biological Sciences, University of Lethbridge, Lethbridge, AB T1K 7X8, Canada; [email protected] * Correspondence: [email protected] (O.K.); [email protected] (I.K.) Simple Summary: In recent years, multiple preclinical studies have shown that changes in endo- cannabinoid system signaling may have various effects on intestinal inflammation and colorectal cancer. However, not all tumors can respond to cannabinoid therapy in the same manner. Given that colorectal cancer is a heterogeneous disease with different genomic landscapes, experiments with cannabinoids should involve different molecular subtypes, emerging mutations, and various stages of the disease. We hope that this review can help researchers form a comprehensive understanding of cannabinoid interactions in colorectal cancer and intestinal bowel diseases. We believe that selecting a particular experimental model based on the disease’s genetic landscape is a crucial step in the drug discovery, which eventually may tremendously benefit patient’s treatment outcomes and bring us one step closer to individualized medicine. Abstract: Despite the multiple preventive measures and treatment options, colorectal cancer holds a significant place in the world’s disease and mortality rates. The development of novel therapy is in Citation: Cherkasova, V.; Kovalchuk, critical need, and based on recent experimental data, cannabinoids could become excellent candidates. O.; Kovalchuk, I. Cannabinoids and This review covered known experimental studies regarding the effects of cannabinoids on intestinal Endocannabinoid System Changes in inflammation and colorectal cancer. In our opinion, because colorectal cancer is a heterogeneous Intestinal Inflammation and disease with different genomic landscapes, the choice of cannabinoids for tumor prevention and Colorectal Cancer. -

Supporting Information
Supporting Information Blankman et al. 10.1073/pnas.1217121110 SI Materials and Methods RT-PCR. Total RNA was isolated from brain tissue of 6-wk-old −/− +/+ Materials. We purchased 2-arachidonoylglycerol (2-AG) from ABHD12 and ABHD12 mice using TRIzol (Invitrogen). Cayman Chemicals; 2-oleoylglycerol, pentadecanoic acid (PDA), First-strand cDNA was synthesized using the SuperScript III and dodecylmonoalkylglycerol ether (C12:0 MAGE) were pur- Reverse Transcriptase kit (Invitrogen) according to the manu- chased from Sigma-Aldrich. Monopentadecanoin (C15:0 MAG) facturer’s protocol. PCR amplification (25 cycles) of a 259 bp fragment of the ABHD12 cDNA was performed with primers 50- and monoheptadecanoin (C17:0 MAG) were purchased from Nu- 0 0 Chek-Prep. Phospholipids and lysophospholipids were purchased CTCAGTAGGAACACCATCGGAGC-3 and 5 -GCCAGGG- AAGTATCGGTATATCACTG-3. Amplification of a 245-bp from Avanti Polar Lipids. Fluorophosphonate (FP)-rhodamine 0 (1) and JZL184 (2) were synthesized as described previously. GAPDH product was performed as a control with primers 5 - GGTGAAGGTCGGTGTGAACGG-30 and 50-CCCATTTGA- 0 Generation of ABHD12−/− Mice. The α/β-hydrolase domain-con- TGTTAGTGGGGTCTCG-3 . Both sets of primers were de- taining (ABHD)12-targeting construct was generated by ampli- signed to span a >2-kb region of genomic DNA. fying 3.4- and 4.4-kb regions of the Abhd12 gene adjacent to the Untargeted liquid chromatography–mass spectrometry proteomic analysis. catalytic exon 8 from a BAC clone containing the Abhd12 locus Mouse brain membrane proteomes were prepared from 6-mo-old − − + + (clone ID RP23-193L22 from BACPAC Resources Center at ABHD12 / and ABHD12 / mice (n = 3 per genotype). -

Cannabinoids in the Pathophysiology of Skin Inflammation
molecules Review Cannabinoids in the Pathophysiology of Skin Inflammation Cristian Scheau 1 , Ioana Anca Badarau 1, Livia-Gratiela Mihai 1, Andreea-Elena Scheau 2, Daniel Octavian Costache 3, Carolina Constantin 4,5, Daniela Calina 6 , Constantin Caruntu 1,7,*, Raluca Simona Costache 8,* and Ana Caruntu 9,10 1 Department of Physiology, “Carol Davila” University of Medicine and Pharmacy, 050474 Bucharest, Romania; [email protected] (C.S.); [email protected] (I.A.B.); [email protected] (L.-G.M.) 2 Department of Radiology and Medical Imaging, Fundeni Clinical Institute, 022328 Bucharest, Romania; [email protected] 3 Department of Dermatology, “Carol Davila” Central Military Emergency Hospital, 010825 Bucharest, Romania; [email protected] 4 Immunology Department, ”Victor Babes” National Institute of Pathology, 050096 Bucharest, Romania; [email protected] 5 Department of Pathology, Colentina University Hospital, 020125 Bucharest, Romania 6 Department of Clinical Pharmacy, University of Medicine and Pharmacy of Craiova, 200349 Craiova, Romania; [email protected] 7 Department of Dermatology, “Prof. N. Paulescu” National Institute of Diabetes, Nutrition and Metabolic Diseases, 011233 Bucharest, Romania 8 Gastroenterology and Internal Medicine Clinic, Carol Davila University Central Emergency Military Hospital, Carol Davila University of Medicine and Pharmacy, 050474 Bucharest, Romania 9 Department of Oral and Maxillofacial Surgery, “Carol Davila” Central Military Emergency Hospital, 010825 Bucharest, Romania; [email protected] 10 Faculty of Medicine, “Titu Maiorescu” University, 031593 Bucharest, Romania * Correspondence: [email protected] (C.C.); [email protected] (R.S.C.); Tel.: +40-745-086-978 (C.C.) Academic Editor: Eric J. Downer Received: 30 December 2019; Accepted: 2 February 2020; Published: 4 February 2020 Abstract: Cannabinoids are increasingly-used substances in the treatment of chronic pain, some neuropsychiatric disorders and more recently, skin disorders with an inflammatory component. -

The Expanded Endocannabinoid System/Endocannabinoidome As a Potential Target for Treating Diabetes Mellitus
Current Diabetes Reports (2019) 19:117 https://doi.org/10.1007/s11892-019-1248-9 OBESITY (KM GADDE, SECTION EDITOR) The Expanded Endocannabinoid System/Endocannabinoidome as a Potential Target for Treating Diabetes Mellitus Alain Veilleux1,2,3 & Vincenzo Di Marzo1,2,3,4,5 & Cristoforo Silvestri3,4,5 # Springer Science+Business Media, LLC, part of Springer Nature 2019 Abstract Purpose of Review The endocannabinoid (eCB) system, i.e. the receptors that respond to the psychoactive component of cannabis, their endogenous ligands and the ligand metabolic enzymes, is part of a larger family of lipid signals termed the endocannabinoidome (eCBome). We summarize recent discoveries of the roles that the eCBome plays within peripheral tissues in diabetes, and how it is being targeted, in an effort to develop novel therapeutics for the treatment of this increasingly prevalent disease. Recent Findings As with the eCB system, many eCBome members regulate several physiological processes, including energy intake and storage, glucose and lipid metabolism and pancreatic health, which contribute to the development of type 2 diabetes (T2D). Preclinical studies increasingly support the notion that targeting the eCBome may beneficially affect T2D. Summary The eCBome is implicated in T2D at several levels and in a variety of tissues, making this complex lipid signaling system a potential source of many potential therapeutics for the treatments for T2D. Keywords Endocannabinoidome . Bioactive lipids . Peripheral tissues . Glucose . Insulin Introduction: The Endocannabinoid System cannabis-derived natural product, Δ9-tetrahydrocannabinol and its Subsequent Expansion (THC), responsible for most of the psychotropic, euphoric to the “Endocannabinoidome” and appetite-stimulating actions (via CB1 receptors) and immune-modulatory effects (via CB2 receptors) of marijuana, The discovery of two G protein-coupled receptors, the canna- opened the way to the identification of the endocannabinoids binoid receptor type-1 (CB1) and − 2 (CB2) [1, 2], for the (eCBs). -
Marinol Cannabidiol C21H30O2 Trade Name
Cannabinoids are a group of terpenophenolic compounds secreted by Cannabis flowers that provide relief from a wide array of symptoms including, pain, nausea, and inflammation. They operate by imitating the body’s natural endocannabinoids, which activate to maintain internal stability and overall health. When consumed, cannabinoids bind to receptor sites throughout the brain (CB1 receptors) and body (CB2 receptors). Different cannabinoids have different effects based on their binding affinity for each receptor. By targeting specific cannabinoids at these receptors, different types of relief can be achieved. Presently, there are at least 113 different cannabinoids isolated from Cannabis—each exhibiting varied effects. THC Tetrahydrocannabinol C21H30O2 Trade name: Marinol Legal Status: US – Schedule I, Schedule II (as Cesamet), Schedule III (as Marinol) OH CA – Schedule II UK – Class B AU – S8 (controlled) H Psychoactive Tetrahydrocannabinol (THC) is typically the most abundant cannabinoid present in cannabis products on the market today. THC has very high psychoactive characteristics and is associated with the ‘high’ and euphoria experienced when using cannabis products. When smoked or ingested, THC binds to cannabinoid receptors throughout the body and affects memory, O coordination, concentration, pleasure, and time perception. H Medicinal Benefits Analgesic • Anti-nauseant • Appetite Stimulant Reduces Glaucoma Symptoms Sleep Aid • Reduces Anxiety and PTSD Symptoms CBD Cannabidiol C21H30O2 Trade name: Epidiolex OH Legal Status: US – Schedule I CA – Schedule II UK – POM (Perscription only) H AU – S4 (Perscription only) non-psychoactive Cannabidiol (CBD) is a major phytocannabinoid and accounts for up to 40% of the plant’s extract. Due to its lack of psychoactivity and HO non-interference with motor and psychological functions, it is a leading candidate for a wide variety of medical applications. -

Recent Developments in Cannabis Chemistry
Recent Developments in Cannabis Chemistry BY ALEXANDER T. SHULGIN, Ph.D. The marijuana plant Cannabis sativa contains a bewildering Introduction array of organic chemicals. As is true with other botanic species, there are representatives of almost all chemical classes present, including mono- and sesquiterpenes, carbohy- drates, aromatics, and a variety of nitrogenous compounds. Interest in the study of this plant has centered primarily on the resinous fraction, as it is this material that is invested with the pharmacological activity that is peculiar to the plant. This resin is secreted by the female plant as a protective agent during seed ripening, although it can be found as a microscopic exudate through the aerial portions of plants of either sex. The pure resin, hashish or charas, is the most potent fraction of the plant, and has served as the source material for most of the chemical studies. The family of chemicals that has been isolated from this source has been referred to as the cannabinoid group. It is unique amongst psychotropic materials from plants in that there are no alkaloids present. The fraction is totally nitro- gen-free. Rather, the set of compounds can be considered as analogs of the parent compound cannabinol (I), a fusion product of terpene and a substituted resorcinol. Beyond the scope of this present review are such questions as the distribution of these compounds within the plant, the bo- tanic variability resulting from geographic distribution, the diversity of pharmacological action assignable to the several Reprinted from Journal of Psychedelic Drugs, vol. II, no. 1, 197 1. 397 398 Marijuana: Medical Papers distinct compounds present, and the various preparations and customs of administration. -

N-Acyl Dopamines Induce Apoptosis in PC12 Cell Line Via the O-1918
bioRxiv preprint doi: https://doi.org/10.1101/075796; this version posted September 19, 2016. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Akimov et al: N-acyl Dopamines Cytotoxicity Receptor N-Acyl Dopamines Induce Apoptosis in PC12 Cell Line via the O-1918-Sensitive non-CB1-non-CB2 Receptor MIKHAIL G. AKIMOV*, ALINA M. ASHBA*, NATALIA M. GRETSKAYA, and VLADIMIR V. BEZUGLOV *: Equally contributed authors Shemyakin-Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, Moscow, Russia Correspondence to: Mikhail G. Akimov, Shemyakin-Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, 117997, ul. Miklukho-Maklaya, 16/10, Moscow, Russia. Tel: +7 4953306592, e-mail: [email protected] Key Words: N-acyl dopamine, cancer, cytotoxicity, apoptosis, non-CB1-non-CB2 receptor. 1 bioRxiv preprint doi: https://doi.org/10.1101/075796; this version posted September 19, 2016. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Akimov et al: N-acyl Dopamines Cytotoxicity Receptor Abstract. Dopamine amides of long chain fatty acids (NADA) are a family of endogenous mammalian lipids with an unknown function; they are anti-proliferative for many cancer cell lines. The aim of this study was to identify the NADA receptor responsible for cell death induction. -

INTRODUCTION One of the Tasks of a Pharmacologist Is the Classification of Drugs
NEUROPSYCHOPHARMACOLOGIC STUDIES OF MARIJUANA: SOME SYNTHETIC AND NATURAL THC DERIVATIVES IN ANIMALS AND MAN* Edward F. Domino Department of Pharmacology, University of Michigan, Ann Arbor 48104 and Lafayette Clinic, Detroit, Mich. 48207 INTRODUCTION One of the tasks of a pharmacologist is the classification of drugs. Marijuana and its derivatives pose some problems. Legally marijuana has been classified as a narcotic, yet pharmacologically it is quite different. Mankind has known about the Cannabis plant from which marijuana and other products are obtained for more than 4,708 years. A considerable body of data is available to describe its gross effects. Over the centuries it has been called a depressant, euphoriant, ine- briant, intoxicant, hallucinogen, psychotomimetic, sedative-hypnotic-anesthetic, and stimulant. Perhaps more than any other drug it is a social irritant that elicits rather remarkable behavior reactions in both users and nonusers. Moreau’ in 1845, employed it to produce a model psychosis, thus initiating the fashion to study hallucinogens as a means of gaining insights into mental illness. Lewin2 classified Cannabis as one of the “phantastica: hallucinating substances.” One text on hallucinogens3 does not even refer to Cannabis, but another does? There are some aspects of the subjective effects of both LSD-25 and Cannabis that are similar, yet the latter produces sedative effects, no significant sympatho- mimetic actions, and no cross-tolerance to LSD-25. Hollister5 reviewed the re- cent findings on the effects in man of marijuana and its putative active ingredient Ag-THC, stressing that current investigators have, for the most part, confirmed the Cannabis-induced clinical syndromes long known to occur. -

SYNTHESIS of 3-HETEROCYCLE PHENYL N-ALKYL CARBAMATES and THEIR ACTIVITY AS FAAH INHIBITORS Doctoral Dissertation
TKK Dissertations 203 Espoo 2009 SYNTHESIS OF 3-HETEROCYCLE PHENYL N-ALKYL CARBAMATES AND THEIR ACTIVITY AS FAAH INHIBITORS Doctoral Dissertation Mikko Myllymäki Helsinki University of Technology Faculty of Chemistry and Materials Sciences Department of Chemistry TKK Dissertations 203 Espoo 2009 SYNTHESIS OF 3-HETEROCYCLE PHENYL N-ALKYL CARBAMATES AND THEIR ACTIVITY AS FAAH INHIBITORS Doctoral Dissertation Mikko Myllymäki Dissertation for the degree of Doctor of Philosophy to be presented with due permission of the Faculty of Chemistry and Materials Sciences for public examination and debate in Auditorium KE2 (Komppa Auditorium) at Helsinki University of Technology (Espoo, Finland) on the 19th of December, 2009, at 12 noon. Helsinki University of Technology Faculty of Chemistry and Materials Sciences Department of Chemistry Teknillinen korkeakoulu Kemian ja materiaalitieteiden tiedekunta Kemian laitos Distribution: Helsinki University of Technology Faculty of Chemistry and Materials Sciences Department of Chemistry P.O. Box 6100 (Kemistintie 1) FI - 02015 TKK FINLAND URL: http://chemistry.tkk.fi/ Tel. +358-9-470 22527 Fax +358-9-470 22538 E-mail: [email protected] © 2009 Mikko Myllymäki ISBN 978-952-248-240-2 ISBN 978-952-248-241-9 (PDF) ISSN 1795-2239 ISSN 1795-4584 (PDF) URL: http://lib.tkk.fi/Diss/2009/isbn9789522482419/ TKK-DISS-2686 Multiprint Oy Espoo 2009 3 AB HELSINKI UNIVERSITY OF TECHNOLOGY ABSTRACT OF DOCTORAL DISSERTATION P.O. BOX 1000, FI-02015 TKK http://www.tkk.fi Author Mikko Juhani Myllymäki Name of the dissertation -

The Use of Cannabinoids in Animals and Therapeutic Implications for Veterinary Medicine: a Review
Veterinarni Medicina, 61, 2016 (3): 111–122 Review Article doi: 10.17221/8762-VETMED The use of cannabinoids in animals and therapeutic implications for veterinary medicine: a review L. Landa1, A. Sulcova2, P. Gbelec3 1Faculty of Medicine, Masaryk University, Brno, Czech Republic 2Central European Institute of Technology, Masaryk University, Brno, Czech Republic 3Veterinary Hospital and Ambulance AA Vet, Prague, Czech Republic ABSTRACT: Cannabinoids/medical marijuana and their possible therapeutic use have received increased atten- tion in human medicine during the last years. This increased attention is also an issue for veterinarians because particularly companion animal owners now show an increased interest in the use of these compounds in veteri- nary medicine. This review sets out to comprehensively summarise well known facts concerning properties of cannabinoids, their mechanisms of action, role of cannabinoid receptors and their classification. It outlines the main pharmacological effects of cannabinoids in laboratory rodents and it also discusses examples of possible beneficial use in other animal species (ferrets, cats, dogs, monkeys) that have been reported in the scientific lit- erature. Finally, the article deals with the prospective use of cannabinoids in veterinary medicine. We have not intended to review the topic of cannabinoids in an exhaustive manner; rather, our aim was to provide both the scientific community and clinical veterinarians with a brief, concise and understandable overview of the use of cannabinoids in veterinary -

The Effects of Cannabinoids on Stress-Coping
Utah State University DigitalCommons@USU All Graduate Theses and Dissertations Graduate Studies 5-2021 The Effects of Cannabinoids on Stress-Coping Behaviors and Neuroendocrinological Measures in Chronic Unpredtictable Stress: A Preclinical Systematic Review and Meta-Analysis Noa Reuveni Utah State University Follow this and additional works at: https://digitalcommons.usu.edu/etd Part of the Psychology Commons Recommended Citation Reuveni, Noa, "The Effects of Cannabinoids on Stress-Coping Behaviors and Neuroendocrinological Measures in Chronic Unpredtictable Stress: A Preclinical Systematic Review and Meta-Analysis" (2021). All Graduate Theses and Dissertations. 8098. https://digitalcommons.usu.edu/etd/8098 This Thesis is brought to you for free and open access by the Graduate Studies at DigitalCommons@USU. It has been accepted for inclusion in All Graduate Theses and Dissertations by an authorized administrator of DigitalCommons@USU. For more information, please contact [email protected]. THE EFFECTS OF CANNABINOIDS ON STRESS-COPING BEHAVIORS AND NEUROENDOCRINOLOGICAL MEASURES IN CHRONIC UNPREDTICTABLE STRESS: A PRECLINICAL SYSTEMATIC REVIEW AND META-ANALYSIS by Noa Reuveni A thesis submitted in partial fulfillment of the requirements for the degree of MASTER OF SCIENCE in Psychology Approved: __________________ __________________ Sara Freeman, Ph.D. Scott Bates, Ph.D. Co-Major Professor Co-Major Professor __________________ __________________ Diana Meter, Ph.D. Sarah Schwartz, Ph.D. Committee Member Committee Member __________________ __________________ Tyler Renshaw, Ph.D. D. Richard Cutler, Ph.D. Committee Member Interim Vice Provost for Graduate Studies UTAH STATE UNIVERSITY Logan, Utah 2021 ii Copyright © Noa Reuveni 2021 All Rights Reserved iii ABSTRACT The Effects of Cannabinoids on Stress-Coping Behaviors and Neuroendocrinological Measures in Chronic Unpredictable Stress: A Preclinical Systematic Review and Meta-Analysis by Noa Reuveni, Master of Science Utah State University, 2021 Major Professors: Dr. -
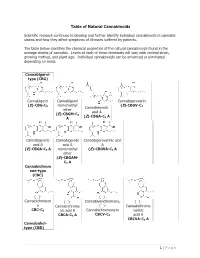
Table of Natural Cannabinoids
Table of Natural Cannabinoids Scientific research continues to develop and further identify individual cannabinoids in cannabis strains and how they affect symptoms of illnesses suffered by patients. The table below identifies the chemical properties of the natural cannabinoids found in the average strains of cannabis. Levels of each of these chemicals will vary with varietal strain, growing method, and plant age. Individual cannabinoids can be enhanced or eliminated depending on need. Cannabigerol- type (CBG) Cannabigerol Cannabigerol Cannabigerovarin (E)-CBG-C monomethyl (E)-CBGV-C 5 Cannabinerolic 3 ether acid A (E)-CBGM-C 5 (Z)-CBGA-C A A 5 Cannabigerolic Cannabigerolic Cannabigerovarinic acid acid A acid A A (E)-CBGA-C5 A monomethyl (E)-CBGVA-C3 A ether (E)-CBGAM- C5 A Cannabichrom ene-type (CBC) (±)- (±)- Cannabichromen (±)- Cannabivarichromene, (±)- e Cannabichrome (±)- Cannabichrome CBC-C5 nic acid A Cannabichromevarin varinic CBCA-C5 A CBCV-C3 acid A CBCVA-C3 A Cannabidiol- type (CBD) 1 | Page (−)-Cannabidiol Cannabidiol Cannabidiol-C4 (−)- Cannabidiorc CBD-C5 momomethyl CBD-C4 Cannabidivarin ol ether CBDV-C3 CBD-C1 CBDM-C5 Cannabidiolic Cannabidivarini acid c acid CBDA-C5 CBDVA-C3 Cannabinodiol- type (CBND) Cannabinodiol Cannabinodivar CBND-C5 in CBND-C3 Tetrahydrocan nabinol-type (THC) 9 9 9 Δ - Δ - Δ - Δ9- Tetrahydrocanna Tetrahydrocan Tetrahydrocannabivarin 9 Tetrahydrocan binol nabinol-C4 Δ -THCV-C3 9 9 nabiorcol Δ -THC-C5 Δ -THC-C4 9 Δ -THCO-C1 9 9 Δ -Tetrahydro- Δ9-Tetrahydro- Δ -Tetrahydro- Δ9-Tetrahydro- cannabinolic