Frances H. Arnold Rudi Fasan, S
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
When the Reaction Is
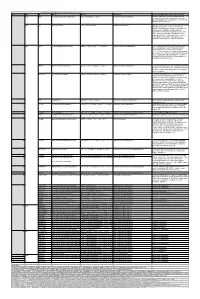
Table S3. iJL1678-ME model modification (blocked reactions) Iter. Cat. ID Name Formula Subsystem Comments (When the reaction is turned on) 1 bp2 EDD 6-phosphogluconate dehydratase 6pgc_c⇌2ddg6p_c + h2o_c Pentose Phosphate Pathway Create a major effect of steep acetate overflow elevation in high growth. Comparing to the main glycolytic pathway, it is metabolicly less efficient but proteomicly more efficient. bp1 ICL Isocitrate lyase icit_c→glx_c + succ_c Anaplerotic Reactions Bypass for the main TCA cycle pathways from turning isocitrate to succinate, when ICL is turned on, Isocitrate dehydrogenase(ICDHyr), 2-Oxogluterate dehydrogenase(AKGDH) and Succinyl-CoA synthetase (ATP-forming,SUCOAS) would reduce. Ref. (1) and (2) shows that this reaction is off in higher growth. Ref. (3) shows that this reaction is converging to being off when the dynamic of respiration using enzyme kinetics is simulated. 2 bp1 ABTA 4-aminobutyrate transaminase 4abut_c + akg_c⇌glu__L_c + sucsal_c Arginine and Proline Metabolism Another backup pathway of succinate production, from 2-Oxoglutarate (akg). Respiration would be induced when it is on, since the flux through ETC(CYTBO3_4pp and ATPS4rpp) would increase. As it requires the co-factor pyridoxal 5'-phosphate(2−) to get catalyzed(4), indicating that this reaction is regulated by the flux of other reactions(pyridoxal 5'- phosphate(2-) production, etc.). 3 GLYAT Glycine C-acetyltransferase accoa_c + gly_c⇌2aobut_c + coa_c Glycine and Serine Metabolism A reaction that back up for the respiration. Reactions fluxes in TCA cycle would drop when this reaction is turned on. It also requires pyridoxal 5'-phosphate(2−) for the regulation. 4 NADTRHD NAD transhydrogenase nad_c + nadph_c⇌nadh_c + nadp_c Oxidative Phosphorylation A reaction that would make the transition between NAD and NADP metabolically more efficient. -

The Human Flavoproteome
CORE Metadata, citation and similar papers at core.ac.uk Provided by Elsevier - Publisher Connector Archives of Biochemistry and Biophysics 535 (2013) 150–162 Contents lists available at SciVerse ScienceDirect Archives of Biochemistry and Biophysics journal homepage: www.elsevier.com/locate/yabbi Review The human flavoproteome ⇑ Wolf-Dieter Lienhart, Venugopal Gudipati, Peter Macheroux Graz University of Technology, Institute of Biochemistry, Petersgasse 12, A-8010 Graz, Austria article info abstract Article history: Vitamin B2 (riboflavin) is an essential dietary compound used for the enzymatic biosynthesis of FMN and Received 17 December 2012 FAD. The human genome contains 90 genes encoding for flavin-dependent proteins, six for riboflavin and in revised form 21 February 2013 uptake and transformation into the active coenzymes FMN and FAD as well as two for the reduction to Available online 15 March 2013 the dihydroflavin form. Flavoproteins utilize either FMN (16%) or FAD (84%) while five human flavoen- zymes have a requirement for both FMN and FAD. The majority of flavin-dependent enzymes catalyze Keywords: oxidation–reduction processes in primary metabolic pathways such as the citric acid cycle, b-oxidation Coenzyme A and degradation of amino acids. Ten flavoproteins occur as isozymes and assume special functions in Coenzyme Q the human organism. Two thirds of flavin-dependent proteins are associated with disorders caused by Folate Heme allelic variants affecting protein function. Flavin-dependent proteins also play an important role in the Pyridoxal 50-phosphate biosynthesis of other essential cofactors and hormones such as coenzyme A, coenzyme Q, heme, pyri- Steroids doxal 50-phosphate, steroids and thyroxine. Moreover, they are important for the regulation of folate Thyroxine metabolites by using tetrahydrofolate as cosubstrate in choline degradation, reduction of N-5.10-meth- Vitamins ylenetetrahydrofolate to N-5-methyltetrahydrofolate and maintenance of the catalytically competent form of methionine synthase. -

(12) Patent Application Publication (10) Pub. No.: US 2017/0137846A1 ATSUMI Et Al
US 2017.0137846A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2017/0137846A1 ATSUMI et al. (43) Pub. Date: May 18, 2017 (54) BACTERIA ENGINEERED FOR Related U.S. Application Data CONVERSION OF ETHYLENE TO (60) Provisional application No. 62/009,857, filed on Jun. N-BUTANOL 9, 2014. (71) Applicant: The Regents of the University of California, Oakland, CA (US) Publication Classification (51) Int. Cl. (72) Inventors: Shota ATSUMI, Davis, CA (US); CI2P 7/06 (2006.01) Michael D. TONEY, Davis, CA (US); CI2P 7/16 (2006.01) Gabriel M. RODRIGUEZ, Davis, CA CI2N 15/52 (2006.01) (US); Yohei TASHIRO, Davis, CA (52) U.S. Cl. (US); Justin B. SIEGEL, Davis, CA CPC ................ CI2P 7/06 (2013.01): CI2N 15/52 (US); D. Alexander CARLIN, Davis, (2013.01); C12Y402/01053 (2013.01); C12Y CA (US); Irina KORYAKINA, Davis, 402/01095 (2013.01); C12Y 114/13 (2013.01); CA (US); Shuchi H. DESAI, Davis, CI2Y 102/01002 (2013.01); C12Y 114/13069 CA (US) (2013.01); C12Y 203/01009 (2013.01); C12Y 101/01157 (2013.01); C12Y402/01 (2013.01); (73) Assignee: The Regents of the University of CI2Y 103/01038 (2013.01); C12Y 102/01003 California, Oakland, CA (US) (2013.01); C12P 7/16 (2013.01) (21) Appl. No.: 15/317,656 (57) ABSTRACT (22) PCT Fed: Jun. 9, 2015 The present disclosure provides recombinant bacteria with elevated production of ethanol and/or n-butanol from eth (86) PCT No.: PCT/US 15/34942 ylene. Methods for the production of the recombinant bac S 371 (c)(1), teria, as well as for use thereof for production of ethanol (2) Date: Dec. -

187124736.Pdf
EVALUATING APOENZYME-COENZYME-SUBSTRATE INTERACTIONS OF METHANE MONOOXYGENASE WITH AN ENGINEERED ACTIVE SITE FOR ELECTRON-HARVESTING: A COMPUTATIONAL STUDY A Thesis by SIKAI ZHANG Submitted to the Office of Graduate and Professional Studies of Texas A&M University in partial fulfillment of the requirements for the degree of MASTER OF SCIENCE Chair of Committee, Sandun Fernando Co-Chair of Committee, R. Karthikeyan Committee Member, Carmen Gomes Head of Department, Steve Searcy December 2017 Major Subject: Biological and Agricultural Engineering Copyright 2017 Sikai Zhang ABSTRACT Energy generation via natural gas is viewed as one of the most promising environmentally friendly solutions for ever-increasing energy demand. Of the several alter- natives available, natural gas-powered fuel cells are considered to be one of the most efficient for producing energy. Biocatalysts, present in methanotrophs, known as methane monooxygenases (MMOs) are well known for their ability to quite effectively activate and oxidize methane at low- temperature. To utilize MMOs effectively in a fuel cell, the enzymes should be directly attached onto the anode. However, there is a knowledge gap on how to attach MMOs to an electrode and once attached the impact of active site modification on enzyme functionality. The overall goal of this work was to computationally evaluate the feasibility of attaching MMOs to a metal electrode and evaluate its functionality using docking and molecular dynamic (MD) simulations. It is surmised that MMOs could be attached to a metal electrode by engineering the active site, i.e., Flavin Adenine Dinucleotide (FAD) coenzyme to attract metal clusters (surfaces) via Fe-S functionalization and such modification will keep the active site functionality unfettered. -

Isolation and Characterization of a Rhodococcus Strain Able to Degrade 2-Fluorophenol
Appl Microbiol Biotechnol (2012) 95:511–520 DOI 10.1007/s00253-011-3696-2 ENVIRONMENTAL BIOTECHNOLOGY Isolation and characterization of a Rhodococcus strain able to degrade 2-fluorophenol Anouk F. Duque & Syed A. Hasan & Vânia S. Bessa & Maria F. Carvalho & Ghufrana Samin & Dick B. Janssen & Paula M. L. Castro Received: 15 October 2011 /Accepted: 31 October 2011 /Published online: 19 November 2011 # Springer-Verlag 2011 Abstract A pure bacterial culture able to utilize 2- Introduction fluorophenol (2-FP) as sole carbon and energy source was isolated by selective enrichment from sediments collected The rapid growth of industrial organic synthesis has led to from a contaminated site in Northern Portugal. 16S rRNA the widespread production and release of xenobiotic gene analysis showed that the organism (strain FP1) compounds, whose fate in biological waste treatment belongs to the genus Rhodococcus. When grown aerobi- processes and in the environment is often problematic and cally on 2-FP, growth kinetics of strain FP1 followed the unknown. Halogenated compounds are frequently detected Luong model. An inhibitory effect of increasing 2-FP in waste streams, and their recalcitrant and toxic properties concentrations was observed with no growth occurring at 2- have stimulated studies on their microbial metabolism and FP levels higher than ca. 4 mM. Rhodococcus strain FP1 mineralization. The biodegradation of many chlorinated was able to degrade a range of other organofluorine organic compounds has been studied (Chaudhry and compounds, including 2-fluorobenzoate, 3-fluorobenzoate, Chapalamadugu 1991; Fetzner 1998; Haggblom 1992; 4-fluorobenzoate, 3-fluorophenol, 4-fluorophenol, 3- Janssen et al. 1994; van Pée and Unversucht 2003). -

Catalytic and Hydrodynamic Properties of Styrene Monooxygenases from Rhodococcus Opacus 1CP Are Modulated by Cofactor Binding
Riedel et al. AMB Expr (2015) 5:30 DOI 10.1186/s13568-015-0112-9 ORIGINAL ARTICLE Open Access Catalytic and hydrodynamic properties of styrene monooxygenases from Rhodococcus opacus 1CP are modulated by cofactor binding Anika Riedel1,2*, Thomas Heine1,2, Adrie H Westphal2, Catleen Conrad1, Philipp Rathsack3, Willem J H van Berkel2 and Dirk Tischler1,2* Abstract Styrene monooxygenases (SMOs) are flavoenzymes catalyzing the epoxidation of styrene into styrene oxide. SMOs are composed of a monooxygenase (StyA) and a reductase (StyB). The latter delivers reduced FAD to StyA on the expense of NADH. We identified Rhodococcus opacus 1CP as the first microorganism to possess three different StyA isoforms occurring in two systems StyA1/StyA2B and StyA/StyB, respectively. The hydrodynamic properties of StyA isozymes were found to be modulated by the binding of the (reduced) FAD cofactor. StyA1 and SyA2B mainly occur as dimers in their active forms while StyA is a monomer. StyA1 showed the highest epoxidation activity and excellent enanti‑ oselectivity in aromatic sulfoxidation. The hydrodynamic and biocatalytic properties of SMOs from strain 1CP are of relevance for investigation of possible industrial applications. Keywords: Flavoprotein, Monooxygenase, Oligomerization, FAD binding, Rhodococcus opacus 1CP, Styrene epoxidation Introduction isolates of various soil samples (Hartmans et al. 1990), Styrene monooxygenases (SMO; EC 1.14.14.11) are two- followed by SMOs from Pseudomonas fluorescens ST component flavoenzymes composed of a monooxyge- (Beltrametti et al. 1997; Gennaro et al. 1999; Marconi nase (StyA) and a FAD reductase (StyB) (Hartmans et al. et al. 1996), Pseudomonas spp. Y2 (Velasco et al. 1998) 1990; Huijbers et al. -

Influence of Lipid Membrane Environment on the Kinetics of the Cytochrome P450 Reductase- Cytochrome P450 3A4 Enzyme System in Nanodiscs
Influence of Lipid Membrane Environment on the Kinetics of the Cytochrome P450 Reductase- Cytochrome P450 3A4 Enzyme System in Nanodiscs A thesis submitted to the University of Manchester for the degree of Doctor of Philosophy (PhD) in the Faculty of Science and Engineering 2016 Kang-cheng Liu School of Chemistry Table of Contents List of Figures 5 List of Tables 8 List of Equations 8 List of Abbreviations 9 Abstracts 13 Declaration 14 Copyright Statement 14 Acknowledgements 15 Chapter 1 Introduction 16 1.1 Cytochrome P450 17 1.1.1 The Discovery and Character of CYP 17 1.1.2 Electron Transfer in CYP Systems 22 1.1.3 CYP Catalytic Reaction 24 1.1.4 Catalytic Cycle of CYP 24 1.2 Cytochrome P450 Reductase 28 1.2.1 Electron Transfer 29 1.2.2 Conformational Changes 30 1.3 Cytochrome P450 3A4 32 1.3.1 Physiological Importance 32 1.3.2 The Structure of CYP3A4 34 1.3.3 Cooperative Behaviour of CYP3A4 Kinetics 35 1.4 Interaction between CYP and CPR 41 1.4.1 The Structural Basis of CYP-CPR Interaction 41 1.4.2 Organisation of CPR-CYPs and CYP-CYPs Complex 44 1.5 Microsomal Lipids and CYP System 46 1.6 Nanodiscs 47 1.6.1 Self-Assembly of Nanodiscs 52 1.6.2 Applications of Nanodiscs 54 1.7 Aims and Objectives 64 Chapter 2 Materials and Methods 65 2.1 Chemicals and Reagents 66 1 2.1.1 Bacterial Strains and Culture Media 68 2.2 Basic Molecular Biology Techniques 69 2.2.1 Plasmid Isolation 69 2.2.2 Agarose Gel Electrophoresis 69 2.2.3 Transformation of E. -

The Human Flavoproteome
Archives of Biochemistry and Biophysics 535 (2013) 150–162 Contents lists available at SciVerse ScienceDirect Archives of Biochemistry and Biophysics journal homepage: www.elsevier.com/locate/yabbi Review The human flavoproteome ⇑ Wolf-Dieter Lienhart, Venugopal Gudipati, Peter Macheroux Graz University of Technology, Institute of Biochemistry, Petersgasse 12, A-8010 Graz, Austria article info abstract Article history: Vitamin B2 (riboflavin) is an essential dietary compound used for the enzymatic biosynthesis of FMN and Received 17 December 2012 FAD. The human genome contains 90 genes encoding for flavin-dependent proteins, six for riboflavin and in revised form 21 February 2013 uptake and transformation into the active coenzymes FMN and FAD as well as two for the reduction to Available online 15 March 2013 the dihydroflavin form. Flavoproteins utilize either FMN (16%) or FAD (84%) while five human flavoen- zymes have a requirement for both FMN and FAD. The majority of flavin-dependent enzymes catalyze Keywords: oxidation–reduction processes in primary metabolic pathways such as the citric acid cycle, b-oxidation Coenzyme A and degradation of amino acids. Ten flavoproteins occur as isozymes and assume special functions in Coenzyme Q the human organism. Two thirds of flavin-dependent proteins are associated with disorders caused by Folate Heme allelic variants affecting protein function. Flavin-dependent proteins also play an important role in the Pyridoxal 50-phosphate biosynthesis of other essential cofactors and hormones such as coenzyme A, coenzyme Q, heme, pyri- Steroids doxal 50-phosphate, steroids and thyroxine. Moreover, they are important for the regulation of folate Thyroxine metabolites by using tetrahydrofolate as cosubstrate in choline degradation, reduction of N-5.10-meth- Vitamins ylenetetrahydrofolate to N-5-methyltetrahydrofolate and maintenance of the catalytically competent form of methionine synthase. -

Cloning of Two Gene Clusters Involved in the Catabolism of 2,4-Dinitrophenol by Paraburkholderia Sp
bioRxiv preprint doi: https://doi.org/10.1101/749879; this version posted August 29, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Cloning of Two Gene Clusters Involved in the Catabolism of 2,4-Dinitrophenol by Paraburkholderia sp. 2 Strain KU-46 and Characterization of the Initial DnpAB Enzymes and a Two-Component 3 Monooxygenase DnpC1C2 4 5 Taisei Yamamoto,a Yaxuan Liu,a Nozomi Kohaya,a Yoshie Hasegawa,a Peter C.K. Lau,b Hiroaki Iwakia# 6 7 aDepartment of Life Science & Biotechnology, Kansai University, Suita, Osaka, Japan 8 bDepartment of Microbiology and Immunology, McGill University, Montréal, Quebec, Canada 9 10 11 Running Head: Unique Pathway of 2,4-Dinitrophenol Catabolism 12 #Address correspondence to Hiroaki Iwaki, [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/749879; this version posted August 29, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 13 Abstract 14 Besides an industrial pollutant, 2,4-dinitrophenol (DNP) has been used illegally as a weight loss drug that 15 had claimed human lives. Little is known about the metabolism of DNP, particularly among 16 Gram-negative bacteria. In this study, two non-contiguous genetic loci of Paraburkholderia (formerly 17 Burkholderia) sp. strain KU-46 genome were identified and four key initial genes (dnpA, dnpB, and 18 dnpC1C2) were characterized to provide molecular and biochemical evidence for the degradation of DNP 19 via the formation of 4-nitrophenol (NP), a pathway that is unique among DNP utilizing bacteria. -

Enzyme Turnover Numbers
Kinetic profiling of metabolic specialists demonstrates stability and consistency of in vivo enzyme turnover numbers David Heckmanna,1, Anaamika Campeaub, Colton J. Lloyda, Patrick V. Phaneufa, Ying Hefnera, Marvic Carrillo-Terrazasb, Adam M. Feista,c, David J. Gonzalezb, and Bernhard O. Palssona,c,1 aDepartment of Bioengineering, University of California San Diego, La Jolla, CA 92093; bDepartment of Pharmacology, Skaggs School of Pharmacy and Pharmaceutical Sciences, University of California San Diego, La Jolla, CA 92093; and cThe Novo Nordisk Foundation Center for Biosustainability, Technical University of Denmark, 2800 Lyngby, Denmark Edited by Athel Cornish-Bowden, Institut de Microbiologie de la Méditerranée, CNRS, Marseille, France, and accepted by Editorial Board Member Alan R. Fersht July 28, 2020 (received for review February 11, 2020) Enzyme turnover numbers (kcats) are essential for a quantitative proteomic quantification techniques are still suffering from high k understanding of cells. Because cats are traditionally measured in variation, and previous estimates of kcat were based on in silico 13 low-throughput assays, they can be inconsistent, labor-intensive to flux predictions rather than C fluxomics data. Furthermore, kcat obtain, and can miss in vivo effects. We use a data-driven approach is expected to scale with growth rate (4). As many experimental k Escherichia coli to estimate in vivo cats using metabolic specialist conditions in the literature data used in Davidi et al. (3) resulted in strains that resulted from -

Domain Dynamics and Control of Electron Flux of NADPH Cytochrome P450 Oxidoreductase
Universidade Nova de Lisboa Instituto de Higiene e Medicina Tropical Domain dynamics and control of electron flux of NADPH cytochrome P450 oxidoreductase Diana Isabel Viana da Fonseca Campelo Delgado DISSERTAÇÃO PARA A OBTENÇÃO DO GRAU DE DOUTOR NO RAMO DE CIÊNCIAS BIOMÉDICAS, ESPECIALIDADE DE GENÉTICA MAIO 2019 Universidade Nova de Lisboa Instituto de Higiene e Medicina Tropical Domain dynamics and control of electron flux of NADPH cytochrome P450 oxidoreductase Autor: Diana Isabel Viana da Fonseca Campelo Delgado Orientador: Michel Kranendonk Co-orientador: Celso Cunha Dissertação apresentada para cumprimento dos requisitos necessários à obtenção do grau de Doutor no Ramo de Ciências Biomédicas, especialidade de Genética Apoio financeiro da Fundação para a Ciência e a Tecnologia (projecto FCT-ANR/BEX- BCM/0002/2013). Dedico este trabalho aos que me são mais queridos, Ao meu marido, aos meus pais, aos meus irmãos e sobrinhos. AGRADECIMENTOS Gostaria de expressar o meu agradecimento e gratidão a todos os que apoiaram e contribuíram para a realização deste longo trabalho. Ao Doutor Michel Kranendonk, agradeço a oportunidade de realizar este estudo sob a sua orientação e todos os ensinamentos ao longo destes anos. Ao Professor Celso Cunha, por ter aceite orientar este trabalho a partir do Instituto de Higiene e Medicina Tropical. Quero agradecer a todos os meus colegas de laboratório que me acompanharam ao longo do doutoramento. Francisco Esteves, Susana Silva, Bruno Gomes, Bernardo Palma, Célia Martins, Cláudio Pinheiro, Rita Jerónimo, Rita Lourenço, Marisa Cardoso, D. Lucrécia. Agradeço o apoio e convívio amigo, que tornaram todas as horas de trabalho mais fáceis. A todos os colegas e colaboradores do ToxOmics. -

(19) United States (12) Patent Application Publication (10) Pub
US 20130244920A1 (19) United States (12) Patent Application Publication (10) Pub. N0.: US 2013/0244920 A1 Lee et al. (43) Pub. Date: Sep. 19, 2013 (54) WATER SOLUBLE COMPOSITIONS (52) US. Cl. INCORPORATING ENZYMES, AND METHOD USPC ......................................... .. 510/392; 264/299 OF MAKING SAME (57) ABSTRACT (76) Inventors: David M. Lee, CroWn Point, IN (US); Jennifer L‘ Sims’ Lowell’ IN (Us) Disclosed herein are Water soluble compositions, such as ?lms, including a mixture of a ?rst Water-soluble resin, an (21) Appl' NO': 13/422’709 enzyme, and an enzyme stabilizer Which comprises a func (22) Filed: Man 16, 2012 tional substrate for the enzyme, methods of making such compositions, and methods of using such compositions, e.g. Publication Classi?cation to make packets containing functional ingredients. The enzymes can include proteases and mixtures of proteases (51) Int. Cl. With other enzymes, and the compositions provide good C11D 3/386 (2006.01) retention of enzyme function following ?lm processing and B29C 39/02 (2006.01) storage. US 2013/0244920 A1 Sep. 19,2013 WATER SOLUBLE COMPOSITIONS preheated to a temperature less than 77° C., optionally in a INCORPORATING ENZYMES, AND METHOD range ofabout 66° C. to about 77° C., or about 74° C.; drying OF MAKING SAME the Water from the cast mixture over a period of less than 24 hours, optionally less than 12 hours, optionally less than 8 FIELD OF THE DISCLOSURE hours, optionally less than 2 hours, optionally less than 1 [0001] The present disclosure relates generally to Water hour, optionally less than 45 minutes, optionally less than 30 soluble ?lms.