187124736.Pdf
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Enzyme DHRS7
Toward the identification of a function of the “orphan” enzyme DHRS7 Inauguraldissertation zur Erlangung der Würde eines Doktors der Philosophie vorgelegt der Philosophisch-Naturwissenschaftlichen Fakultät der Universität Basel von Selene Araya, aus Lugano, Tessin Basel, 2018 Originaldokument gespeichert auf dem Dokumentenserver der Universität Basel edoc.unibas.ch Genehmigt von der Philosophisch-Naturwissenschaftlichen Fakultät auf Antrag von Prof. Dr. Alex Odermatt (Fakultätsverantwortlicher) und Prof. Dr. Michael Arand (Korreferent) Basel, den 26.6.2018 ________________________ Dekan Prof. Dr. Martin Spiess I. List of Abbreviations 3α/βAdiol 3α/β-Androstanediol (5α-Androstane-3α/β,17β-diol) 3α/βHSD 3α/β-hydroxysteroid dehydrogenase 17β-HSD 17β-Hydroxysteroid Dehydrogenase 17αOHProg 17α-Hydroxyprogesterone 20α/βOHProg 20α/β-Hydroxyprogesterone 17α,20α/βdiOHProg 20α/βdihydroxyprogesterone ADT Androgen deprivation therapy ANOVA Analysis of variance AR Androgen Receptor AKR Aldo-Keto Reductase ATCC American Type Culture Collection CAM Cell Adhesion Molecule CYP Cytochrome P450 CBR1 Carbonyl reductase 1 CRPC Castration resistant prostate cancer Ct-value Cycle threshold-value DHRS7 (B/C) Dehydrogenase/Reductase Short Chain Dehydrogenase Family Member 7 (B/C) DHEA Dehydroepiandrosterone DHP Dehydroprogesterone DHT 5α-Dihydrotestosterone DMEM Dulbecco's Modified Eagle's Medium DMSO Dimethyl Sulfoxide DTT Dithiothreitol E1 Estrone E2 Estradiol ECM Extracellular Membrane EDTA Ethylenediaminetetraacetic acid EMT Epithelial-mesenchymal transition ER Endoplasmic Reticulum ERα/β Estrogen Receptor α/β FBS Fetal Bovine Serum 3 FDR False discovery rate FGF Fibroblast growth factor HEPES 4-(2-Hydroxyethyl)-1-Piperazineethanesulfonic Acid HMDB Human Metabolome Database HPLC High Performance Liquid Chromatography HSD Hydroxysteroid Dehydrogenase IC50 Half-Maximal Inhibitory Concentration LNCaP Lymph node carcinoma of the prostate mRNA Messenger Ribonucleic Acid n.d. -

Noelia Díaz Blanco
Effects of environmental factors on the gonadal transcriptome of European sea bass (Dicentrarchus labrax), juvenile growth and sex ratios Noelia Díaz Blanco Ph.D. thesis 2014 Submitted in partial fulfillment of the requirements for the Ph.D. degree from the Universitat Pompeu Fabra (UPF). This work has been carried out at the Group of Biology of Reproduction (GBR), at the Department of Renewable Marine Resources of the Institute of Marine Sciences (ICM-CSIC). Thesis supervisor: Dr. Francesc Piferrer Professor d’Investigació Institut de Ciències del Mar (ICM-CSIC) i ii A mis padres A Xavi iii iv Acknowledgements This thesis has been made possible by the support of many people who in one way or another, many times unknowingly, gave me the strength to overcome this "long and winding road". First of all, I would like to thank my supervisor, Dr. Francesc Piferrer, for his patience, guidance and wise advice throughout all this Ph.D. experience. But above all, for the trust he placed on me almost seven years ago when he offered me the opportunity to be part of his team. Thanks also for teaching me how to question always everything, for sharing with me your enthusiasm for science and for giving me the opportunity of learning from you by participating in many projects, collaborations and scientific meetings. I am also thankful to my colleagues (former and present Group of Biology of Reproduction members) for your support and encouragement throughout this journey. To the “exGBRs”, thanks for helping me with my first steps into this world. Working as an undergrad with you Dr. -
When the Reaction Is
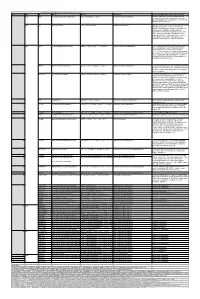
Table S3. iJL1678-ME model modification (blocked reactions) Iter. Cat. ID Name Formula Subsystem Comments (When the reaction is turned on) 1 bp2 EDD 6-phosphogluconate dehydratase 6pgc_c⇌2ddg6p_c + h2o_c Pentose Phosphate Pathway Create a major effect of steep acetate overflow elevation in high growth. Comparing to the main glycolytic pathway, it is metabolicly less efficient but proteomicly more efficient. bp1 ICL Isocitrate lyase icit_c→glx_c + succ_c Anaplerotic Reactions Bypass for the main TCA cycle pathways from turning isocitrate to succinate, when ICL is turned on, Isocitrate dehydrogenase(ICDHyr), 2-Oxogluterate dehydrogenase(AKGDH) and Succinyl-CoA synthetase (ATP-forming,SUCOAS) would reduce. Ref. (1) and (2) shows that this reaction is off in higher growth. Ref. (3) shows that this reaction is converging to being off when the dynamic of respiration using enzyme kinetics is simulated. 2 bp1 ABTA 4-aminobutyrate transaminase 4abut_c + akg_c⇌glu__L_c + sucsal_c Arginine and Proline Metabolism Another backup pathway of succinate production, from 2-Oxoglutarate (akg). Respiration would be induced when it is on, since the flux through ETC(CYTBO3_4pp and ATPS4rpp) would increase. As it requires the co-factor pyridoxal 5'-phosphate(2−) to get catalyzed(4), indicating that this reaction is regulated by the flux of other reactions(pyridoxal 5'- phosphate(2-) production, etc.). 3 GLYAT Glycine C-acetyltransferase accoa_c + gly_c⇌2aobut_c + coa_c Glycine and Serine Metabolism A reaction that back up for the respiration. Reactions fluxes in TCA cycle would drop when this reaction is turned on. It also requires pyridoxal 5'-phosphate(2−) for the regulation. 4 NADTRHD NAD transhydrogenase nad_c + nadph_c⇌nadh_c + nadp_c Oxidative Phosphorylation A reaction that would make the transition between NAD and NADP metabolically more efficient. -

The Human Flavoproteome
CORE Metadata, citation and similar papers at core.ac.uk Provided by Elsevier - Publisher Connector Archives of Biochemistry and Biophysics 535 (2013) 150–162 Contents lists available at SciVerse ScienceDirect Archives of Biochemistry and Biophysics journal homepage: www.elsevier.com/locate/yabbi Review The human flavoproteome ⇑ Wolf-Dieter Lienhart, Venugopal Gudipati, Peter Macheroux Graz University of Technology, Institute of Biochemistry, Petersgasse 12, A-8010 Graz, Austria article info abstract Article history: Vitamin B2 (riboflavin) is an essential dietary compound used for the enzymatic biosynthesis of FMN and Received 17 December 2012 FAD. The human genome contains 90 genes encoding for flavin-dependent proteins, six for riboflavin and in revised form 21 February 2013 uptake and transformation into the active coenzymes FMN and FAD as well as two for the reduction to Available online 15 March 2013 the dihydroflavin form. Flavoproteins utilize either FMN (16%) or FAD (84%) while five human flavoen- zymes have a requirement for both FMN and FAD. The majority of flavin-dependent enzymes catalyze Keywords: oxidation–reduction processes in primary metabolic pathways such as the citric acid cycle, b-oxidation Coenzyme A and degradation of amino acids. Ten flavoproteins occur as isozymes and assume special functions in Coenzyme Q the human organism. Two thirds of flavin-dependent proteins are associated with disorders caused by Folate Heme allelic variants affecting protein function. Flavin-dependent proteins also play an important role in the Pyridoxal 50-phosphate biosynthesis of other essential cofactors and hormones such as coenzyme A, coenzyme Q, heme, pyri- Steroids doxal 50-phosphate, steroids and thyroxine. Moreover, they are important for the regulation of folate Thyroxine metabolites by using tetrahydrofolate as cosubstrate in choline degradation, reduction of N-5.10-meth- Vitamins ylenetetrahydrofolate to N-5-methyltetrahydrofolate and maintenance of the catalytically competent form of methionine synthase. -

Project List 2020
Nottingham BBSRC DTP Project List 2020 For interview candidates Contents Introduction ............................................................................................................................................ 2 Ageing...................................................................................................................................................... 3 Animal Health .......................................................................................................................................... 5 Animal Welfare ........................................................................................................................................ 7 Bioenergy ................................................................................................................................................ 7 Chemical Biology ..................................................................................................................................... 8 Crop Science .......................................................................................................................................... 10 Diet and Health ..................................................................................................................................... 11 Immunology .......................................................................................................................................... 12 Industrial Biotechnology ....................................................................................................................... -

Relating Metatranscriptomic Profiles to the Micropollutant
1 Relating Metatranscriptomic Profiles to the 2 Micropollutant Biotransformation Potential of 3 Complex Microbial Communities 4 5 Supporting Information 6 7 Stefan Achermann,1,2 Cresten B. Mansfeldt,1 Marcel Müller,1,3 David R. Johnson,1 Kathrin 8 Fenner*,1,2,4 9 1Eawag, Swiss Federal Institute of Aquatic Science and Technology, 8600 Dübendorf, 10 Switzerland. 2Institute of Biogeochemistry and Pollutant Dynamics, ETH Zürich, 8092 11 Zürich, Switzerland. 3Institute of Atmospheric and Climate Science, ETH Zürich, 8092 12 Zürich, Switzerland. 4Department of Chemistry, University of Zürich, 8057 Zürich, 13 Switzerland. 14 *Corresponding author (email: [email protected] ) 15 S.A and C.B.M contributed equally to this work. 16 17 18 19 20 21 This supporting information (SI) is organized in 4 sections (S1-S4) with a total of 10 pages and 22 comprises 7 figures (Figure S1-S7) and 4 tables (Table S1-S4). 23 24 25 S1 26 S1 Data normalization 27 28 29 30 Figure S1. Relative fractions of gene transcripts originating from eukaryotes and bacteria. 31 32 33 Table S1. Relative standard deviation (RSD) for commonly used reference genes across all 34 samples (n=12). EC number mean fraction bacteria (%) RSD (%) RSD bacteria (%) RSD eukaryotes (%) 2.7.7.6 (RNAP) 80 16 6 nda 5.99.1.2 (DNA topoisomerase) 90 11 9 nda 5.99.1.3 (DNA gyrase) 92 16 10 nda 1.2.1.12 (GAPDH) 37 39 6 32 35 and indicates not determined. 36 37 38 39 S2 40 S2 Nitrile hydration 41 42 43 44 Figure S2: Pearson correlation coefficients r for rate constants of bromoxynil and acetamiprid with 45 gene transcripts of ECs describing nucleophilic reactions of water with nitriles. -

Bacterial Oxygen Production in the Dark
HYPOTHESIS AND THEORY ARTICLE published: 07 August 2012 doi: 10.3389/fmicb.2012.00273 Bacterial oxygen production in the dark Katharina F. Ettwig*, Daan R. Speth, Joachim Reimann, Ming L. Wu, Mike S. M. Jetten and JanT. Keltjens Department of Microbiology, Institute for Water and Wetland Research, Radboud University Nijmegen, Nijmegen, Netherlands Edited by: Nitric oxide (NO) and nitrous oxide (N2O) are among nature’s most powerful electron Boran Kartal, Radboud University, acceptors. In recent years it became clear that microorganisms can take advantage of Netherlands the oxidizing power of these compounds to degrade aliphatic and aromatic hydrocar- Reviewed by: bons. For two unrelated bacterial species, the “NC10” phylum bacterium “Candidatus Natalia Ivanova, Lawrence Berkeley National Laboratory, USA Methylomirabilis oxyfera” and the γ-proteobacterial strain HdN1 it has been suggested Carl James Yeoman, Montana State that under anoxic conditions with nitrate and/or nitrite, monooxygenases are used for University, USA methane and hexadecane oxidation, respectively. No degradation was observed with *Correspondence: nitrous oxide only. Similarly, “aerobic” pathways for hydrocarbon degradation are employed − Katharina F.Ettwig, Department of by (per)chlorate-reducing bacteria, which are known to produce oxygen from chlorite (ClO ). Microbiology, Institute for Water and 2 Wetland Research, Radboud In the anaerobic methanotroph M. oxyfera, which lacks identifiable enzymes for nitrogen University Nijmegen, formation, substrate activation in the presence of nitrite was directly associated with both Heyendaalseweg 135, 6525 AJ oxygen and nitrogen formation. These findings strongly argue for the role of NO, or an Nijmegen, Netherlands. e-mail: [email protected] oxygen species derived from it, in the activation reaction of methane. -

Bacterial Degradation of Isoprene in the Terrestrial Environment Myriam
Bacterial Degradation of Isoprene in the Terrestrial Environment Myriam El Khawand A thesis submitted to the University of East Anglia in fulfillment of the requirements for the degree of Doctor of Philosophy School of Environmental Sciences November 2014 ©This copy of the thesis has been supplied on condition that anyone who consults it is understood to recognise that its copyright rests with the author and that use of any information derive there from must be in accordance with current UK Copyright Law. In addition, any quotation or extract must include full attribution. Abstract Isoprene is a climate active gas emitted from natural and anthropogenic sources in quantities equivalent to the global methane flux to the atmosphere. 90 % of the emitted isoprene is produced enzymatically in the chloroplast of terrestrial plants from dimethylallyl pyrophosphate via the methylerythritol pathway. The main role of isoprene emission by plants is to reduce the damage caused by heat stress through stabilizing cellular membranes. Isoprene emission from microbes, animals, and humans has also been reported, albeit less understood than isoprene emission from plants. Despite large emissions, isoprene is present at low concentrations in the atmosphere due to its rapid reactions with other atmospheric components, such as hydroxyl radicals. Isoprene can extend the lifetime of potent greenhouse gases, influence the tropospheric concentrations of ozone, and induce the formation of secondary organic aerosols. While substantial knowledge exists about isoprene production and atmospheric chemistry, our knowledge of isoprene sinks is limited. Soils consume isoprene at a high rate and contain numerous isoprene-utilizing bacteria. However, Rhodococcus sp. AD45 is the only terrestrial isoprene-degrading bacterium characterized in any detail. -

Integrative Structure Modeling: Overview and Assessment
BI88CH06_Kalisman ARjats.cls May 17, 2019 13:46 Annual Review of Biochemistry Integrative Structure Modeling: Overview and Assessment Merav Braitbard,1 Dina Schneidman-Duhovny,1,2 and Nir Kalisman1 1Department of Biological Chemistry, Institute of Life Sciences, The Hebrew University of Jerusalem, Jerusalem 9190401, Israel; email: [email protected] 2School of Computer Science and Engineering, The Hebrew University of Jerusalem, Jerusalem 9190401, Israel; email: [email protected] Annu. Rev. Biochem. 2019. 88:113–35 Keywords First published as a Review in Advance on integrative modeling, macromolecular assemblies, protein structure, March 4, 2019 cross-linking, mass spectrometry, cryo–electron microscopy, cryo-EM The Annual Review of Biochemistry is online at biochem.annualreviews.org Abstract https://doi.org/10.1146/annurev-biochem-013118- Integrative structure modeling computationally combines data from multi- Access provided by Howard University on 11/09/19. For personal use only. 111429 ple sources of information with the aim of obtaining structural insights that Annu. Rev. Biochem. 2019.88:113-135. Downloaded from www.annualreviews.org Copyright © 2019 by Annual Reviews. are not revealed by any single approach alone. In the first part of this re- All rights reserved view, we survey the commonly used sources of structural information and the computational aspects of model building. Throughout the past decade, integrative modeling was applied to various biological systems, with a focus on large protein complexes. Recent progress in the field of cryo–electron mi- croscopy (cryo-EM) has resolved many of these complexes to near-atomic resolution. In the second part of this review, we compare a range of pub- lished integrative models with their higher-resolution counterparts with the aim of critically assessing their accuracy. -

(12) Patent Application Publication (10) Pub. No.: US 2017/0137846A1 ATSUMI Et Al
US 2017.0137846A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2017/0137846A1 ATSUMI et al. (43) Pub. Date: May 18, 2017 (54) BACTERIA ENGINEERED FOR Related U.S. Application Data CONVERSION OF ETHYLENE TO (60) Provisional application No. 62/009,857, filed on Jun. N-BUTANOL 9, 2014. (71) Applicant: The Regents of the University of California, Oakland, CA (US) Publication Classification (51) Int. Cl. (72) Inventors: Shota ATSUMI, Davis, CA (US); CI2P 7/06 (2006.01) Michael D. TONEY, Davis, CA (US); CI2P 7/16 (2006.01) Gabriel M. RODRIGUEZ, Davis, CA CI2N 15/52 (2006.01) (US); Yohei TASHIRO, Davis, CA (52) U.S. Cl. (US); Justin B. SIEGEL, Davis, CA CPC ................ CI2P 7/06 (2013.01): CI2N 15/52 (US); D. Alexander CARLIN, Davis, (2013.01); C12Y402/01053 (2013.01); C12Y CA (US); Irina KORYAKINA, Davis, 402/01095 (2013.01); C12Y 114/13 (2013.01); CA (US); Shuchi H. DESAI, Davis, CI2Y 102/01002 (2013.01); C12Y 114/13069 CA (US) (2013.01); C12Y 203/01009 (2013.01); C12Y 101/01157 (2013.01); C12Y402/01 (2013.01); (73) Assignee: The Regents of the University of CI2Y 103/01038 (2013.01); C12Y 102/01003 California, Oakland, CA (US) (2013.01); C12P 7/16 (2013.01) (21) Appl. No.: 15/317,656 (57) ABSTRACT (22) PCT Fed: Jun. 9, 2015 The present disclosure provides recombinant bacteria with elevated production of ethanol and/or n-butanol from eth (86) PCT No.: PCT/US 15/34942 ylene. Methods for the production of the recombinant bac S 371 (c)(1), teria, as well as for use thereof for production of ethanol (2) Date: Dec. -

Methane Copper Peroxo Complexes and Structural Studies on Transition Metal Bis(Pyrazolyl)Methane Complexes
Dissertation zur Erlangung des Doktorgrades der Fakultät für Chemie und Pharmazie der Ludwig-Maximilians-Universität München Biomimetic hydroxylation catalysis with bis(pyrazolyl)methane copper peroxo complexes and structural studies on transition metal bis(pyrazolyl)methane complexes Claudia Gerlinde Wilfer aus Dachau 2015 Erklärung Diese Dissertation wurde im Sinne von §7 der Promotionsordnung vom 28. November 2011 von Frau Prof. Dr. Herres-Pawlis betreut. Eidesstattliche Versicherung Diese Dissertation wurde eigenständig und ohne unerlaubte Hilfe erarbeitet. München, den 23.02.2015 ................................................................... Dissertation eingereicht am 24.02.2015 1. Gutachterin: Prof. Dr. Sonja Herres-Pawlis 2. Gutachter: Prof. Dr. Peter Klüfers Mündliche Prüfung am 26.03.2015 Kurzzusammenfassung Im Hinblick auf die Entwicklung biomimetischer Tyrosinasemodelle wurde im Rahmen dieser Arbeit HC(3-tBuPz) 2(1-MeIm) als Bis(pyrazolyl)methan-Ligand zweiter Generation entwickelt, 2 2 der in der Lage ist, den Cu 2O2-Peroxo-Komplex (µ-η :η )-[Cu 2O2{HC(3-tBuPz) 2 (1-MeIm)} 2][SbF 6]2 zu stabilisieren. Er konnte sowohl UV/Vis- und röntgenabsorptionsspektroskopisch als auch massenspektrometrisch nachgewiesen werden. Hierbei wurde eine Blau-Verschiebung der für Komplexe dieser Art bekannten Charge- Transfer-Banden beobachtet. Dieser Effekt wurde mit dichtefunktionaltheoretischen Methoden untersucht und zeigte eine starke Donorfähigkeit der Imidazolyl-Einheit des Liganden als Ursache. Es wurde festgestellt, dass der Komplex -

Inactivation of the Particulate Methane Monooxygenase (Pmmo) in Methylo- Coccus Capsulatus (Bath) by Acetylene
ÔØ ÅÒÙ×Ö ÔØ Inactivation of the particulate methane monooxygenase (pMMO) in Methylo- coccus capsulatus (Bath) by acetylene Minh D. Pham, Ya-Ping Lin, Quan Van Vuong, Penumaka Nagababu, Brian T.-A. Chang, Kok Yaoh Ng, Chein-Hung Chen, Chau-Chung Han, Chung-Hsuan Chen, Mai Suan Li, Steve S.-F. Yu, Sunney I. Chan PII: S1570-9639(15)00208-3 DOI: doi: 10.1016/j.bbapap.2015.08.004 Reference: BBAPAP 39641 To appear in: BBA - Proteins and Proteomics Received date: 16 June 2015 Revised date: 29 July 2015 Accepted date: 9 August 2015 Please cite this article as: Minh D. Pham, Ya-Ping Lin, Quan Van Vuong, Penumaka Nagababu, Brian T.-A. Chang, Kok Yaoh Ng, Chein-Hung Chen, Chau-Chung Han, Chung-Hsuan Chen, Mai Suan Li, Steve S.-F. Yu, Sunney I. Chan, Inactivation of the particulate methane monooxygenase (pMMO) in Methylococcus capsulatus (Bath) by acetylene, BBA - Proteins and Proteomics (2015), doi: 10.1016/j.bbapap.2015.08.004 This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain. ACCEPTED MANUSCRIPT Inactivation of the particulate methane monooxygenase (pMMO) in Methylococcus capsulatus (Bath) by acetylene☆ Minh D. Phama,b,c, Ya-Ping Lind, Quan Van Vuonge, Penumaka Nagababua, Brian T.-A.