Lung-Specific Inactivation of C/Ebpα Causes a Pathological Pattern Characteristic of COPD
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Prospective Isolation of NKX2-1–Expressing Human Lung Progenitors Derived from Pluripotent Stem Cells
The Journal of Clinical Investigation RESEARCH ARTICLE Prospective isolation of NKX2-1–expressing human lung progenitors derived from pluripotent stem cells Finn Hawkins,1,2 Philipp Kramer,3 Anjali Jacob,1,2 Ian Driver,4 Dylan C. Thomas,1 Katherine B. McCauley,1,2 Nicholas Skvir,1 Ana M. Crane,3 Anita A. Kurmann,1,5 Anthony N. Hollenberg,5 Sinead Nguyen,1 Brandon G. Wong,6 Ahmad S. Khalil,6,7 Sarah X.L. Huang,3,8 Susan Guttentag,9 Jason R. Rock,4 John M. Shannon,10 Brian R. Davis,3 and Darrell N. Kotton1,2 2 1Center for Regenerative Medicine, and The Pulmonary Center and Department of Medicine, Boston University School of Medicine, Boston, Massachusetts, USA. 3Center for Stem Cell and Regenerative Medicine, Brown Foundation Institute of Molecular Medicine, University of Texas Health Science Center, Houston, Texas, USA. 4Department of Anatomy, UCSF, San Francisco, California, USA. 5Division of Endocrinology, Diabetes and Metabolism, Beth Israel Deaconess Medical Center and Harvard Medical School, Boston, Massachusetts, USA. 6Department of Biomedical Engineering and Biological Design Center, Boston University, Boston, Massachusetts, USA. 7Wyss Institute for Biologically Inspired Engineering, Harvard University, Boston, Massachusetts, USA. 8Columbia Center for Translational Immunology & Columbia Center for Human Development, Columbia University Medical Center, New York, New York, USA. 9Department of Pediatrics, Monroe Carell Jr. Children’s Hospital, Vanderbilt University, Nashville, Tennessee, USA. 10Division of Pulmonary Biology, Cincinnati Children’s Hospital, Cincinnati, Ohio, USA. It has been postulated that during human fetal development, all cells of the lung epithelium derive from embryonic, endodermal, NK2 homeobox 1–expressing (NKX2-1+) precursor cells. -

Deciphering the Nature of Trp73 Isoforms in Mouse
cancers Article Deciphering the Nature of Trp73 Isoforms in Mouse Embryonic Stem Cell Models: Generation of Isoform-Specific Deficient Cell Lines Using the CRISPR/Cas9 Gene Editing System Lorena López-Ferreras 1,2,†, Nicole Martínez-García 1,3,†, Laura Maeso-Alonso 1,2,‡, Marta Martín-López 1,4,‡, Ángela Díez-Matilla 1,‡ , Javier Villoch-Fernandez 1,2, Hugo Alonso-Olivares 1,2, Margarita M. Marques 3,5,* and Maria C. Marin 1,2,* 1 Instituto de Biomedicina (IBIOMED), Universidad de León, 24071 León, Spain; [email protected] (L.L.-F.); [email protected] (N.M.-G.); [email protected] (L.M.-A.); [email protected] (M.M.-L.); [email protected] (Á.D.-M.); [email protected] (J.V.-F.); [email protected] (H.A.-O.) 2 Departamento de Biología Molecular, Universidad de León, 24071 León, Spain 3 Departamento de Producción Animal, Universidad de León, 24071 León, Spain 4 Biomar Microbial Technologies, Parque Tecnológico de León, Armunia, 24009 León, Spain 5 Instituto de Desarrollo Ganadero y Sanidad Animal (INDEGSAL), Universidad de León, 24071 León, Spain * Correspondence: [email protected] (M.M.M.); [email protected] (M.C.M.); Tel.: +34-987-291757 Citation: López-Ferreras, L.; (M.M.M.); +34-987-291490 (M.C.M.) Martínez-García, N.; Maeso-Alonso, † Equal contribution. ‡ Equal contribution. L.; Martín-López, M.; Díez-Matilla, Á.; Villoch-Fernandez, J.; Simple Summary: The Trp73 gene is involved in the regulation of multiple biological processes Alonso-Olivares, H.; Marques, M.M.; Marin, M.C. Deciphering the Nature such as response to stress, differentiation and tissue architecture. -

Ten Commandments for a Good Scientist
Unravelling the mechanism of differential biological responses induced by food-borne xeno- and phyto-estrogenic compounds Ana María Sotoca Covaleda Wageningen 2010 Thesis committee Thesis supervisors Prof. dr. ir. Ivonne M.C.M. Rietjens Professor of Toxicology Wageningen University Prof. dr. Albertinka J. Murk Personal chair at the sub-department of Toxicology Wageningen University Thesis co-supervisor Dr. ir. Jacques J.M. Vervoort Associate professor at the Laboratory of Biochemistry Wageningen University Other members Prof. dr. Michael R. Muller, Wageningen University Prof. dr. ir. Huub F.J. Savelkoul, Wageningen University Prof. dr. Everardus J. van Zoelen, Radboud University Nijmegen Dr. ir. Toine F.H. Bovee, RIKILT, Wageningen This research was conducted under the auspices of the Graduate School VLAG Unravelling the mechanism of differential biological responses induced by food-borne xeno- and phyto-estrogenic compounds Ana María Sotoca Covaleda Thesis submitted in fulfillment of the requirements for the degree of doctor at Wageningen University by the authority of the Rector Magnificus Prof. dr. M.J. Kropff, in the presence of the Thesis Committee appointed by the Academic Board to be defended in public on Tuesday 14 September 2010 at 4 p.m. in the Aula Unravelling the mechanism of differential biological responses induced by food-borne xeno- and phyto-estrogenic compounds. Ana María Sotoca Covaleda Thesis Wageningen University, Wageningen, The Netherlands, 2010, With references, and with summary in Dutch. ISBN: 978-90-8585-707-5 “Caminante no hay camino, se hace camino al andar. Al andar se hace camino, y al volver la vista atrás se ve la senda que nunca se ha de volver a pisar” - Antonio Machado – A mi madre. -

C/Ebpα Is an Essential Collaborator in Hoxa9/Meis1-Mediated Leukemogenesis
C/EBPα is an essential collaborator in Hoxa9/Meis1-mediated leukemogenesis Cailin Collinsa, Jingya Wanga, Hongzhi Miaoa, Joel Bronsteina, Humaira Nawera, Tao Xua, Maria Figueroaa, Andrew G. Munteana, and Jay L. Hessa,b,1 aDepartment of Pathology, University of Michigan, Ann Arbor, MI 48109; and bIndiana University School of Medicine, Indianapolis, IN 46202 Edited* by Louis M. Staudt, National Institutes of Health, Bethesda, MD, and approved May 19, 2014 (received for review February 12, 2014) Homeobox A9 (HOXA9) is a homeodomain-containing transcrip- with Hoxa9. In addition, C/EBP recognition motifs are enriched tion factor that plays a key role in hematopoietic stem cell expan- at Hoxa9 binding sites. sion and is commonly deregulated in human acute leukemias. A C/EBPα is a basic leucine-zipper transcription factor that plays variety of upstream genetic alterations in acute myeloid leukemia a critical role in lineage commitment during hematopoietic dif- −/− (AML) lead to overexpression of HOXA9, almost always in associ- ferentiation (18). Whereas Cebpa mice show complete loss of ation with overexpression of its cofactor meis homeobox 1 (MEIS1). the granulocytic compartment, recent work shows that loss of α A wide range of data suggests that HOXA9 and MEIS1 play a syn- C/EBP in adult HSCs leads to both an increase in the number ergistic causative role in AML, although the molecular mechanisms of functional HSCs and an increase in their proliferative and leading to transformation by HOXA9 and MEIS1 remain elusive. In repopulating capacity (19, 20). Conversely, CEBPA overexpression can promote transdifferentiation of a variety of fibroblastic cells to this study, we identify CCAAT/enhancer binding protein alpha (C/ the myeloid lineage and can induce monocytic differentiation in EBPα) as a critical collaborator required for Hoxa9/Meis1-mediated α MLL-fusion protein-mediated leukemias (21, 22). -

Chloride Channels Regulate Differentiation and Barrier Functions
RESEARCH ARTICLE Chloride channels regulate differentiation and barrier functions of the mammalian airway Mu He1†*, Bing Wu2†, Wenlei Ye1, Daniel D Le2, Adriane W Sinclair3,4, Valeria Padovano5, Yuzhang Chen6, Ke-Xin Li1, Rene Sit2, Michelle Tan2, Michael J Caplan5, Norma Neff2, Yuh Nung Jan1,7,8, Spyros Darmanis2*, Lily Yeh Jan1,7,8* 1Department of Physiology, University of California, San Francisco, San Francisco, United States; 2Chan Zuckerberg Biohub, San Francisco, United States; 3Department of Urology, University of California, San Francisco, San Francisco, United States; 4Division of Pediatric Urology, University of California, San Francisco, Benioff Children’s Hospital, San Francisco, United States; 5Department of Cellular and Molecular Physiology, Yale University School of Medicine, New Heaven, United States; 6Department of Anesthesia and Perioperative Care, University of California, San Francisco, San Francisco, United States; 7Department of Biochemistry and Biophysics, University of California, San Francisco, San Francisco, United States; 8Howard Hughes Medical Institute, University of California, San Francisco, San Francisco, United States *For correspondence: Abstract The conducting airway forms a protective mucosal barrier and is the primary target of [email protected] (MH); [email protected] airway disorders. The molecular events required for the formation and function of the airway (SD); mucosal barrier, as well as the mechanisms by which barrier dysfunction leads to early onset airway [email protected] (LYJ) diseases, -

Test Summary Flyer-NGS Panels.Pub
Cancer‐related Mutaon Analysis Next Generaon Gene Sequencing for Myeloid Tesng Assay Summary IU Health Molecular Pathology Laboratory now offers high throughput sequencing for hot spot mutations found in clinically relevant cancer genes. In addition to a general panel of 54 genes, selected panels have been developed for a more tai- lored application in specific cancers. Comparing to single gene assay, these panels offer a more comprehensive and eco- nomic way to assess prognosis and/or treatment options for cancer patients at the initial diagnosis or at the relapse. Orderable Name: Use IU Health Molecular Pathology requisition; Call 317.491.6417 for requisition. Panels include: AML Mutations by NGS ASXL1, CEBPA, DNMT3A, ETV6/TEL, FLT3, HRAS, IDH1, IDH2, KIT, KRAS, MLL, NPM1, NRAS, PHF6, RUNX1, TET2, TP53, WT1 MDS Mutations by NGS ASXL1, ATRX, BCOR, BCORL1, ETV6/TEL, DNMT3A, EZH2, GNAS, IDH1, IDH2, RUNX1, SF3B1, SRSF2, TET2, TP53, U2AF1, ZRSR2 CML Mutations by NGS ABL1 MPN Mutations by NGS ASXL1, BRAF, CALR, CSF3R, EZH2, IKZF1, JAK2, JAK3, KDM6A, KIT, MPL, PDGRA, SETBP1, TET2 CMML Mutations by NGS ASXL1, CBL, CBLB, CBLC, EZH2, RUNX1, TET2, TP53, SRSF2 JMML Mutations by NGS CBL, CBLB, CBLC, HRAS, KRAS, NRAS, PTPN11 ALL Mutations by NGS ABL1, CSF3R, FBXW7, IKZF1, JAK3, KDM6A, NOTCH1 CLL Mutations by NGS MYD88, NOTCH1, SF3B1, TP53 Lymphoma/Myeloma Mutations by NGS BRAF, CDKN2A, CSF3R, FBXW7, HRAS, KRAS, MYD88, NOTCH1, NRAS, SF3B1,TP53 Hematopoietic Neoplasms Mutations by NGS ABL1, ASXL1, ATRX, BCOR, BCORL1, BRAF, CALR, CBL, CBLB, CBLC,CDKN2A, CEBPA, CSF3R, CUX1, DNMT3A, ETV6/TEL, EZH2, FBXW7, FLT3, GATA1, GATA2, GNAS, HRAS, IDH1, IDH2, IKZF1,JAK2, JAK3, KDM6A, KIT, KRAS, MLL, MPL, MYD88, NOTCH1, NPM1, NRAS, PDGFRA, PHF6, PTEN, PTPN11, RAD21, RUNX1,SETBP1, SF3B1, SMC1A, SMC3, SRSF2, STAG2, TET2, TP53, U2AF1, WT1, ZRSR2 Clinical Utility: This test is useful for the assessment of prognosis and/or treatment options for cancer patients at the initial diagnosis or at the relapse. -
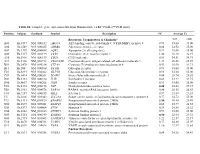
Table S1. Complete Gene Expression Data from Human Diabetes RT² Profiler™ PCR Array Receptors, Transporters & Channels* A
Table S1. Complete gene expression data from Human Diabetes RT² Profiler™ PCR Array Position Unigene GenBank Symbol Description FC Average Ct Receptors, Transporters & Channels* NGT GDM A01 Hs,5447 NM_000352 ABCC8 ATP-binding cassette, sub-family C (CFTR/MRP), member 8 0.93 35.00 35.00 A04 0Hs,2549 NM_000025 ADRB3 Adrenergic, beta-3-, receptor 0.88 34.92 35.00 A07 Hs,1307 NM_000486 AQP2 Aquaporin 2 (collecting duct) 0.93 35.00 35.00 A09 30Hs,5117 NM_001123 CCR2 Chemokine (C-C motif) receptor 2 1.00 26.28 26.17 A10 94Hs,5916 396NM_006139 CD28 CD28 molecule 0.81 34.51 34.71 A11 29Hs,5126 NM_001712 CEACAM1 Carcinoembryonic antigen-related cell adhesion molecule 1 1.31 26.08 25.59 B01 82Hs,2478 NM_005214 CTLA4 (biliaryCytotoxic glycoprotein) T-lymphocyte -associated protein 4 0.53 30.90 31.71 B11 24Hs,208 NM_000160 GCGR Glucagon receptor 0.93 35.00 35.00 C01 Hs,3891 NM_002062 GLP1R Glucagon-like peptide 1 receptor 0.93 35.00 35.00 C07 03Hs,6434 NM_000201 ICAM1 Intercellular adhesion molecule 1 0.84 28.74 28.89 D02 47Hs,5134 NM_000418 IL4R Interleukin 4 receptor 0.64 34.22 34.75 D06 57Hs,4657 NM_000208 INSR Insulin receptor 0.93 35.00 35.00 E05 44Hs,4312 NM_006178 NSF N-ethylmaleimide-sensitive factor 0.48 28.42 29.37 F08 79Hs,2961 NM_004578 RAB4A RAB4A, member RAS oncogene family 0.88 20.55 20.63 F10 69Hs,7287 NM_000655 SELL Selectin L 0.97 23.89 23.83 F11 56Hs,3806 NM_001042 SLC2A4 Solute carrier family 2 (facilitated glucose transporter), member 4 0.77 34.72 35.00 F12 91Hs,5111 NM_003825 SNAP23 Synaptosomal-associated protein, 23kDa 3.90 -

KRAS Drives Immune Evasion in a Genetic Model of Pancreatic Cancer
ARTICLE https://doi.org/10.1038/s41467-021-21736-w OPEN KRAS drives immune evasion in a genetic model of pancreatic cancer Irene Ischenko1, Stephen D’Amico1, Manisha Rao2, Jinyu Li2, Michael J. Hayman1, Scott Powers 2, ✉ ✉ Oleksi Petrenko 1,3 & Nancy C. Reich 1,3 Immune evasion is a hallmark of KRAS-driven cancers, but the underlying causes remain unresolved. Here, we use a mouse model of pancreatic ductal adenocarcinoma to inactivate 1234567890():,; KRAS by CRISPR-mediated genome editing. We demonstrate that at an advanced tumor stage, dependence on KRAS for tumor growth is reduced and is manifested in the sup- pression of antitumor immunity. KRAS-deficient cells retain the ability to form tumors in immunodeficient mice. However, they fail to evade the host immune system in syngeneic wild-type mice, triggering strong antitumor response. We uncover changes both in tumor cells and host immune cells attributable to oncogenic KRAS expression. We identify BRAF and MYC as key mediators of KRAS-driven tumor immune suppression and show that loss of BRAF effectively blocks tumor growth in mice. Applying our results to human PDAC we show that lowering KRAS activity is likewise associated with a more vigorous immune environment. 1 Department of Molecular Genetics and Microbiology, Stony Brook University, Stony Brook, NY, USA. 2 Department of Pathology, Stony Brook University, ✉ Stony Brook, NY, USA. 3These authors jointly supervised this work: Oleksi Petrenko, Nancy C. Reich. email: [email protected]; [email protected] NATURE COMMUNICATIONS | (2021) 12:1482 | https://doi.org/10.1038/s41467-021-21736-w | www.nature.com/naturecommunications 1 ARTICLE NATURE COMMUNICATIONS | https://doi.org/10.1038/s41467-021-21736-w RAS is frequently associated with some of the deadliest and characterization of KRASG12D p53KO mouse cell lines forms of cancer. -

Genetic Testing for Acute Myeloid Leukemia AHS-M2062
Corporate Medical Policy Genetic Testing for Acute Myeloid Leukemia AHS-M2062 File Name: genetic_testing_for_acute_myeloid_leukemia Origination: 1/1/2019 Last CAP Review: 8/2021 Next CAP Review: 8/2022 Last Review: 8/2021 Description of Procedure or Service Acute myeloid leukemia (AML) is characterized by large numbers of abnormal, immature myeloid cells in the bone marrow and peripheral blood resulting from genetic changes in hematopoietic precursor cells which disrupt normal hematopoietic growth and differentiation (Stock, 2020). Related Policies: Genetic Cancer Susceptibility Using Next Generation Sequencing AHS-M2066 Molecular Panel Testing of Cancers to Identify Targeted Therapy AHS-M2109 Serum Tumor Markers for Malignancies AHS-G2124 Minimal Residual Disease (MRD) AHS- M2175 ***Note: This Medical Policy is complex and technical. For questions concerning the technical language and/or specific clinical indications for its use, please consult your physician. Policy BCBSNC will provide coverage for genetic testing for acute myeloid leukemia when it is determined to be medically necessary because the medical criteria and guidelines shown below are met. Benefits Application This medical policy relates only to the services or supplies described herein. Please refer to the Member's Benefit Booklet for availability of benefits. Member's benefits may vary according to benefit design; therefore member benefit language should be reviewed before applying the terms of this medical policy. When Genetic Testing for Acute Myeloid Leukemia is covered The use of genetic testing for acute myeloid leukemia is considered medically necessary for the following: A. Genetic testing for FLT3 internal tandem duplication and tyrosine kinase domain mutations (ITD and TKD), IDH1, IDH2, TET2, WT1, DNMT3A, ASXL1 and/or TP53 in adult and pediatric patients with suspected or confirmed AML of any type for prognostic and/or therapeutic purposes. -

Rb Family Proteins Differentially Regulate Distinct Cell Lineages During Epithelial Development Kathryn A
Research article 4299 Rb family proteins differentially regulate distinct cell lineages during epithelial development Kathryn A. Wikenheiser-Brokamp Department of Pathology and Immunology, and Division of Molecular Oncology in the Department of Internal Medicine, Washington University School of Medicine, 660 South Euclid Avenue, St Louis, MO 63110, USA e-mail: [email protected] Accepted 23 April 2004 Development 131, 4299-4310 Published by The Company of Biologists 2004 doi:10.1242/dev.01232 Summary pRb, p107 and p130 are important regulators of cell cycle cell types. Ablation of total Rb family function resulted and have extensive overlapping functions; however, only in opposing effects in specification along distinct cell Rb has been shown to be a bone fide tumor suppressor. lineages, providing evidence that pocket proteins inhibit Defining the overlapping versus distinct pocket protein neuroendocrine cell fate while being required for functions is therefore an important step to understanding differentiation in other cell types. These findings identify a the unique role of Rb. Using lung as a model, the present novel role for pocket proteins in cell fate determination, studies demonstrate that pocket proteins are important not and establish a unique cell lineage-specific function for Rb only in regulating cell cycle and survival but also in cell that explains, at least in part, why Rb and p16 are lineage specification. An inducible lung-specific Rb inactivated in phenotypically distinct carcinomas. knockout strategy was used to demonstrate that Rb is specifically required for restricting neuroendocrine cell fate Key words: Rb, p107, p130, Cre-LoxP system, Lung development, despite functional compensation for Rb deficiency in other CC10 Introduction overexpressed (Classon and Dyson, 2001; Harbour and Dean, The retinoblastoma protein (Rb) is a prototype tumor 2000). -

Regulation of the C/Ebpα Signaling Pathway in Acute Myeloid Leukemia (Review)
ONCOLOGY REPORTS 33: 2099-2106, 2015 Regulation of the C/EBPα signaling pathway in acute myeloid leukemia (Review) GUANHUA SONG1, LIn Wang2, Kehong BI3 and guosheng JIang1 1Department of hemato-oncology, Institute of Basic Medicine, shandong academy of Medical sciences, Key Laboratory for Modern Medicine and Technology of Shandong Province, Key Laboratory for Rare and Uncommon Diseases, Key Medical Laboratory for Tumor Immunology and Traditional Chinese Medicine Immunology of shandong Province, Jinan, Shandong 250062; 2Research Center for Medical Biotechnology, Shandong Academy of Medical Sciences, Jinan, Shandong 250062; 3Department of Hematology, Qianfoshan Mountain Hospital of Shandong University, Jinan, Shandong 250014, P.R. China Received December 2, 2014; Accepted January 26, 2015 DoI: 10.3892/or.2015.3848 Abstract. The transcription factor CCAAT/enhancer binding Contents protein α (C/EBPα), as a critical regulator of myeloid devel- opment, directs granulocyte and monocyte differentiation. 1. Introduction Various mechanisms have been identified to explain how 2. Function of C/EBPα in myeloid differentiation C/EBPα functions in patients with acute myeloid leukemia 3. Regulation of the C/EBPα signaling pathway (AML). C/EBPα expression is suppressed as a result of 4. Conclusion common leukemia-associated genetic and epigenetic altera- tions such as AML1-ETO, RARα-PLZF or gene promoter methylation. Recent data have shown that ubiquitination modi- 1. Introduction fication also contributes to its downregulation. In addition, 10-15% of patients with AML in an intermediate cytogenetic Acute myeloid leukemia (AML) is characterized by uncon- risk subgroup were characterized by mutations of the C/EBPα trolled proliferation of myeloid progenitors that exhibit a gene. -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in