A Gene-Based Positive Selection Detection Approach to Identify Vaccine Candidates Using Toxoplasma Gondii As a Test Case Protozoan Pathogen
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Plasmodium Falciparum Is Not As Lonely As Previously Considered
AUTOPHAGIC PUNCTUM ARTICLE ADDENDUM Virulence 2:1, 71-76; January/February 2011; © 2011 Landes Bioscience Plasmodium falciparum is not as lonely as previously considered Franck Prugnolle,1,* Francisco Ayala,2 Benjamin Ollomo,3 Céline Arnathau,1 Patrick Durand1 and François Renaud1,* 1Laboratoire MIVEGEC; UM1-CNRS 5290-IRD 224, IRD Montpellier, France; 2Department of Ecology and Evolutionary Biology; University of California; Irvine, CA USA; 3Centre International de Recherches Médicales de Franceville; Franceville, Gabon ntil very recently, only one species The identification of Plasmodium spe- U(P. reichenowi) was known to be a cies circulating in great apes in Africa phylogenetic sister lineage of P. falciparum, was primarily done during the first half the main malignant agent of human of the twentieth century, on the basis of malaria. In 2009 and 2010, new studies morphological features.1 This approach have revealed the existence of several new has several limitations.4 First, phenotypic phylogenetic species related to this deadly plasticity can lead to incorrect identifica- parasite and infecting chimpanzees and tions. Second, morphological keys are gorillas in Africa. These discoveries invite often effective only for a particular life us to explore a whole set of new questions, stage which cannot always be observed which we briefly do in this article. or is difficult to be. Finally, and perhaps most important, this approach overlooks The Plasmodium species infecting morphologically cryptic taxa. These limi- humans and non-human primates cluster tations, together with the difficulty to into two distinct phylogenetic lineages collect and manipulate great apes, were (Fig. 1). One of these lineages (in yellow certainly, at least in part, responsible for in Fig. -

Legionella Genus Genome Provide Multiple, Independent Combinations for Replication in Human Cells
Supplemental Material More than 18,000 effectors in the Legionella genus genome provide multiple, independent combinations for replication in human cells Laura Gomez-Valero1,2, Christophe Rusniok1,2, Danielle Carson3, Sonia Mondino1,2, Ana Elena Pérez-Cobas1,2, Monica Rolando1,2, Shivani Pasricha4, Sandra Reuter5+, Jasmin Demirtas1,2, Johannes Crumbach1,2, Stephane Descorps-Declere6, Elizabeth L. Hartland4,7,8, Sophie Jarraud9, Gordon Dougan5, Gunnar N. Schroeder3,10, Gad Frankel3, and Carmen Buchrieser1,2,* Table S1: Legionella strains analyzed in the present study Table S2: Type IV secretion systems predicted in the genomes analyzed Table S3: Eukaryotic like domains identified in the Legionella proteins analyzed Table S4: Small GTPases domains detected in the genus Legionella as defined in the CDD ncbi domain database Table S5: Eukaryotic like proteins detected in the Legionella genomes analyzed in this study Table S6: Aminoacid identity of the Dot/Icm components in Legionella species with respect to orthologous proteins in L. pneumophila Paris Table S7: Distribution of seventeen highly conserved Dot/Icm secreted substrates Table S8: Comparison of the effector reperotoire among strains of the same Legionella species Table S9. Number of Dot/Icm secreted proteins predicted in each strain analyzed Table S10: Replication capacity of the different Legionella species analyzed in this study and collection of literature data on Legionella replication Table S11: Orthologous table for all genes of the 80 analyzed strains based on PanOCT. The orthologoss where defined with the program PanOCT using the parameters previously indicated in material and methods.) Figure S1: Distribution of the genes predicted to encode for the biosynthesis of flagella among all Legionella species. -
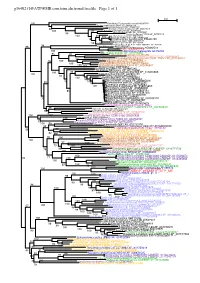
Page 1 G16482.T1@[email protected]
g16482.t1@[email protected] Page 1 of 1 0.2 100 Candidatus Thiomargarita nelsonii KHD07353 Bacillus thuringiensis Bt407 YP_006926419 Legionella pneumophila WP_014842957 88 Endozoicomonas montiporae WP_034879478 Acinetobacter baumannii WP_031968362 100 87 Vibrio nigripulchritudo WP_004402447 98 Psychrobacter sp. P11G3 WP_057760119 100 99 Xenorhabdus bovienii WP_038203911 100 Rouxiella chamberiensis WP_045046329 18 Dendroctonus ponderosae ERL83390 Serratia sp. M24T3 WP_009636677 98 56 100Lonsdalea quercina WP_026743324 Escherichia coli str. K-12 substr. MG1655 NP_415996 86 98 Chlorobi bacterium OLB5 KXK50071 CoptotermesTric_AAA92714 formosanus AGM32510 100 Histomonas meleagridis ACI16482 100Eucalyptus grandis XP_010033838 95 98 Ectocarpus siliculosus CBJ25596 Guillardia theta CCMP2712 XP_005838347 Phaeodactylum tricornutum CCAP 1055/1 XP_002180331 99 Aphanomyces astaci XP_009825348 95 100 Albugo laibachii Nc14 CCA15917 100 Phytophthora parasitica P1569 ETI54450 100 Phytophthora infestans T30-4 XP_002896607 100 Brugia malayi CDP93853 Bombus impatiens XP_003487345 Trichoplax adhaerens XP_002116081 95 Apteryx australis mantelli XP_013810665 100 100 Callorhinchus milii XP_007891153 Homo sapiens AAC50613 93 Callorhinchus milii XP_007900551 100 Clupea harengus XP_012674444 99 Chrysochloris asiatica XP_006865016 82 76 100 84Nannospalax galili XP_008842357 98 Cricetulus griseus XP_007626683 100 65Rousettus aegyptiacus XP_016015809 Condylura cristata XP_004683330 58Chinchilla lanigera XP_013367523 85 61Microcebus murinus XP_012594174 -

Inferring Natural Selection Signals in Plasmodium Vivax-Encoded Proteins Having a Potential Role in Merozoite Invasion
Identificación de señales de selección natural en genes de Plasmodium vivax que codifican proteínas involucradas en el proceso de invasión para determinar su potencial uso en una vacuna antimalárica. Diego Edison Garzón Ospina Tesis Doctoral presentada como requisito para optar al título de Doctor en Ciencias Biomédicas y Biológicas de la Universidad del Rosario Bogotá, 2018 1 Identificación de señales de selección natural en genes de Plasmodium vivax que codifican proteínas involucradas en el proceso de invasión para determinar su potencial uso en una vacuna antimalárica. Estudiante Diego Edison Garzón Ospina Biólogo, Universidad INCCA de Colombia Magister en Ciencias-Microbiología, Universidad Nacional de Colombia Director Manuel Alfonso Patarroyo Gutiérrez M.D., Dr.Sc. Jefe del Departamento de Biología Molecular e Inmunología Fundación Instituto de Inmunología de Colombia (FIDIC) Profesor Titular, Escuela de Medicina y Ciencias de la Salud Universidad del Rosario DOCTORADO EN CIENCIAS BIOMÉDICAS Y BIOLÓGICAS UNIVERSIDAD DEL ROSARIO Bogotá, 2018 2 AGRADECIMIENTOS Quiero dar mis agradecimientos y dedicar este trabajo a mi madre: Martha Ospina Vargas, y a mis hermanos: Yamile Garzón Ospina y Julián David Escobar Ospina, quienes me han acompañado y apoyado en todo momento. A Sindy Paola Buitrago Puentes por su acompañamiento y apoyo durante este tiempo. También agradecer al Dr. Manuel Alfonso Patarroyo Gutiérrez, por haberme acogido en su equipo de trabajo y por apoyarme a lo largo de todos estos años. Quiero agradecer y reconocer la labor de: Andrea Estefanía Ramos, Darwin Andrés Moreno Pérez, Elizabeth Gutiérrez Vásquez, Heidy Daniela Ortiz Suarez, Lady Johanna Forero Rodríguez, Laura Alejandra Ricaurte Contreras, Leidy Paola Reyes, Luis Alfredo Baquero, Paola Andrea Camargo Ayala, Sindy Paola Buitrago Puentes, Ricardo De León Montero y Yimara Grosso Paz, quienes aportaron su tiempo, dedicación y esfuerzo, permitiendo la culminación de este trabajo. -

Debugging Parasite Genomes: Using Metabolic Modeling to Accelerate Antiparasitic Drug Development
Debugging parasite genomes: Using metabolic modeling to accelerate antiparasitic drug development Maureen A. Carey Charlottesville, Virginia Bachelors of Science, Lafayette College 2014 A Dissertation presented to the Graduate Faculty of the University of Virginia in Candidacy for the Degree of Doctor of Philosophy Department of Microbiology, Immunology, and Cancer Biology University of Virginia September, 2018 i M. A. Carey ii Abstract: Eukaryotic parasites, like the casual agent of malaria, kill over one million people around the world annually. Developing novel antiparasitic drugs is a pressing need because there are few available therapeutics and the parasites have developed drug resistance. However, novel drug targets are challenging to identify due to poor genome annotation and experimental challenges associated with growing these parasites. Here, we focus on computational and experimental approaches that generate high-confidence hypotheses to accelerate labor-intensive experimental work and leverage existing experimental data to generate new drug targets. We generate genome-scale metabolic models for over 100 species to develop a parasite knowledgebase and apply these models to contextualize experimental data and to generate candidate drug targets. M. A. Carey iii Figure 0.1: Image from blog.wellcome.ac.uk/2010/06/15/of-parasitology-and-comics/. Preamble: Eukaryotic single-celled parasites cause diseases, such as malaria, African sleeping sickness, diarrheal disease, and leishmaniasis, with diverse clinical presenta- tions and large global impacts. These infections result in over one million preventable deaths annually and contribute to a significant reduction in disability-adjusted life years. This global health burden makes parasitic diseases a top priority of many economic development and health advocacy groups. -

(PAD) and Post-Translational Protein Deimination—Novel Insights Into Alveolata Metabolism, Epigenetic Regulation and Host–Pathogen Interactions
biology Article Peptidylarginine Deiminase (PAD) and Post-Translational Protein Deimination—Novel Insights into Alveolata Metabolism, Epigenetic Regulation and Host–Pathogen Interactions Árni Kristmundsson 1,*, Ásthildur Erlingsdóttir 1 and Sigrun Lange 2,* 1 Institute for Experimental Pathology at Keldur, University of Iceland, Keldnavegur 3, 112 Reykjavik, Iceland; [email protected] 2 Tissue Architecture and Regeneration Research Group, School of Life Sciences, University of Westminster, London W1W 6UW, UK * Correspondence: [email protected] (Á.K.); [email protected] (S.L.) Simple Summary: Alveolates are a major group of free living and parasitic organisms; some of which are serious pathogens of animals and humans. Apicomplexans and chromerids are two phyla belonging to the alveolates. Apicomplexans are obligate intracellular parasites; that cannot complete their life cycle without exploiting a suitable host. Chromerids are mostly photoautotrophs as they can obtain energy from sunlight; and are considered ancestors of the apicomplexans. The pathogenicity and life cycle strategies differ significantly between parasitic alveolates; with some causing major losses in host populations while others seem harmless to the host. As the life cycles of Citation: Kristmundsson, Á.; Erlingsdóttir, Á.; Lange, S. some are still poorly understood, a better understanding of the factors which can affect the parasitic Peptidylarginine Deiminase (PAD) alveolates’ life cycles and survival is of great importance and may aid in new biomarker discovery. and Post-Translational Protein This study assessed new mechanisms relating to changes in protein structure and function (so-called Deimination—Novel Insights into “deimination” or “citrullination”) in two key parasites—an apicomplexan and a chromerid—to Alveolata Metabolism, Epigenetic assess the pathways affected by this protein modification. -

Highly Rearranged Mitochondrial Genome in Nycteria Parasites (Haemosporidia) from Bats
Highly rearranged mitochondrial genome in Nycteria parasites (Haemosporidia) from bats Gregory Karadjiana,1,2, Alexandre Hassaninb,1, Benjamin Saintpierrec, Guy-Crispin Gembu Tungalunad, Frederic Arieye, Francisco J. Ayalaf,3, Irene Landaua, and Linda Duvala,3 aUnité Molécules de Communication et Adaptation des Microorganismes (UMR 7245), Sorbonne Universités, Muséum National d’Histoire Naturelle, CNRS, CP52, 75005 Paris, France; bInstitut de Systématique, Evolution, Biodiversité (UMR 7205), Sorbonne Universités, Muséum National d’Histoire Naturelle, CNRS, Université Pierre et Marie Curie, CP51, 75005 Paris, France; cUnité de Génétique et Génomique des Insectes Vecteurs (CNRS URA3012), Département de Parasites et Insectes Vecteurs, Institut Pasteur, 75015 Paris, France; dFaculté des Sciences, Université de Kisangani, BP 2012 Kisangani, Democratic Republic of Congo; eLaboratoire de Biologie Cellulaire Comparative des Apicomplexes, Faculté de Médicine, Université Paris Descartes, Inserm U1016, CNRS UMR 8104, Cochin Institute, 75014 Paris, France; and fDepartment of Ecology and Evolutionary Biology, University of California, Irvine, CA 92697 Contributed by Francisco J. Ayala, July 6, 2016 (sent for review March 18, 2016; reviewed by Sargis Aghayan and Georges Snounou) Haemosporidia parasites have mostly and abundantly been de- and this lack of knowledge limits the understanding of the scribed using mitochondrial genes, and in particular cytochrome evolutionary history of Haemosporidia, in particular their b (cytb). Failure to amplify the mitochondrial cytb gene of Nycteria basal diversification. parasites isolated from Nycteridae bats has been recently reported. Nycteria parasites have been primarily described, based on Bats are hosts to a diverse and profuse array of Haemosporidia traditional taxonomy, in African insectivorous bats of two fami- parasites that remain largely unstudied. -

A Highly Rearranged Mitochondrial Genome in Nycteria Parasites
A highly rearranged mitochondrial genome in Nycteria parasites (Haemosporidia) from bats Gregory Karadjian, Alexandre Hassanin, Benjamin Saintpierre, Guy-Crispin Gembu Tungaluna, Frederic Ariey, Francisco J. Ayala, Irene Landau, Linda Duval To cite this version: Gregory Karadjian, Alexandre Hassanin, Benjamin Saintpierre, Guy-Crispin Gembu Tungaluna, Fred- eric Ariey, et al.. A highly rearranged mitochondrial genome in Nycteria parasites (Haemosporidia) from bats. Proceedings of the National Academy of Sciences of the United States of America , National Academy of Sciences, 2016, 113 (35), pp.9834 - 9839. 10.1073/pnas.1610643113. hal-01395176 HAL Id: hal-01395176 https://hal.sorbonne-universite.fr/hal-01395176 Submitted on 10 Nov 2016 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution - NonCommercial| 4.0 International License A highly rearranged mitochondrial genome in Nycteria parasites (Haemosporidia) from bats Gregory Karadjian a, b, 1, Alexandre Hassaninc, 1, Benjamin Saintpierred, Guy-Crispin Gembu -

Structural and Functional Insights Into Apicomplexan Gliding and Its Regulation
Structural and functional insights into apicomplexan gliding and its regulation Dissertation to obtain the degree of Doctor of Natural Sciences University of Hamburg Faculty of Mathematics, Informatics and Natural Sciences at the Department of Biology by Samuel Pažický from Bratislava, Slovakia Hamburg 2020 Examination commission Examination commission chair Prof. Dr. Jörg Ganzhorn (University of Hamburg) Examination commission members Prof. Jonas Schmidt-Chanasit (Bernhard Nocht Institute for Tropical Medicine and University of Hamburg) Prof. Tim Gilberger (Bernhard Nocht Institute for Tropical Medicine, Centre for Structural Systems Biology and University of Hamburg) Dr. Maria Garcia-Alai (European Molecular Biology Laboratory and Centre for Structural Systems Biology) Dr. Christian Löw (European Molecular Biology Laboratory and Centre for Structural Systems Biology) Date of defence: 29.01.2021 This work was performed at European Molecular Biology Laboratory, Hamburg Unit under the supervision of Dr. Christian Löw and Prof. Tim-Wolf Gilberger. The work was supported by the Joachim Herz Foundation. Evaluation Prof. Dr. rer. nat. Tim-Wolf Gilberger Bernhard Nocht Institute for Tropical Medicine (BNITM) Department of Cellular Parasitology Hamburg Dr. Christian Löw European Molecular Biology Laboratory Hamburg unit Hamburg Prof. Dr. vet. med. Thomas Krey Hannover Medical School Institute of Virology Declaration of academic honesty I hereby declare, on oath, that I have written the present dissertation by my own and have not used other than the acknowledged resources and aids. Eidesstattliche Erklärung Hiermit erkläre ich an Eides statt, dass ich die vorliegende Dissertationsschrift selbst verfasst und keine anderen als die angegebenen Quellen und Hilfsmittel benutzt habe. Hamburg, 22.9.2020 Samuel Pažický List of contents Declaration of academic honesty 4 List of contents 5 Acknowledgements 6 Summary 7 Zusammenfassung 10 List of publications 12 Scientific contribution to the manuscript 14 Abbreviations 16 1. -

Diversity of Malaria Parasites in Great Apes in Gabon
Boundenga et al. Malaria Journal (2015) 14:111 DOI 10.1186/s12936-015-0622-6 RESEARCH Open Access Diversity of malaria parasites in great apes in Gabon Larson Boundenga1,3*, Benjamin Ollomo1†, Virginie Rougeron1,2, Lauriane Yacka Mouele1, Bertrand Mve-Ondo1, Lucrèce M Delicat-Loembet1, Nancy Diamella Moukodoum1, Alain Prince Okouga1, Céline Arnathau2, Eric Elguero2, Patrick Durand2, Florian Liégeois1,4, Vanina Boué1, Peggy Motsch1, Guillaume Le Flohic1, Alphonse Ndoungouet1, Christophe Paupy1,2, Cheikh Tidiane Ba3, Francois Renaud2 and Franck Prugnolle1,2† Abstract Background: Until 2009, the Laverania subgenus counted only two representatives: Plasmodium falciparum and Plasmodium reichenowi. The recent development of non-invasive methods allowed re-exploration of plasmodial diversity in African apes. Although a large number of great ape populations have now been studied regarding Plasmodium infections in Africa, there are still vast areas of their distribution that remained unexplored. Gabon constitutes an important part of the range of western central African great ape subspecies (Pan troglodytes troglodytes and Gorilla gorilla gorilla), but has not been studied so far. In the present study, the diversity of Plasmodium species circulating in great apes in Gabon was analysed. Methods: The analysis of 1,261 faecal samples from 791 chimpanzees and 470 gorillas collected from 24 sites all over Gabon was performed. Plasmodium infections were characterized by amplification and sequencing of a portion of the Plasmodium cytochrome b gene. Results: The analysis of the 1,261 samples revealed that at least six Plasmodium species circulate in great apes in Gabon (Plasmodium praefalciparum, Plasmodium gorA (syn Plasmodium adleri), Plasmodium gorB (syn Plasmodium blacklocki) in gorillas and Plasmodium gaboni, P. -

The Origins of Malaria: There Are More Things in Heaven and Earth
SUPPLEMENT ARTICLE S16 The origins of malaria: there are more things in heaven and earth ... P. J. KEELING1 and J. C. RAYNER2* 1 Department of Botany, Canadian Institute for Advanced Research, Evolutionary Biology Program, University of British Columbia, Vancouver, BC V6T 1Z4, Canada 2 Malaria Programme, Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge CB10 1SA, UK (Received 9 December 2013; revised 14 April 2014; accepted 15 April 2014; first published online 25 June 2014) SUMMARY Malaria remains one of the most significant global public health burdens, with nearly half of the world’s population at risk of infection. Malaria is not however a monolithic disease – it can be caused by multiple different parasite species of the Plasmodium genus, each of which can induce different symptoms and pathology, and which pose quite different challenges for control. Furthermore, malaria is in no way restricted to humans. There are Plasmodium species that have adapted to infect most warm-blooded vertebrate species, and the genus as a whole is both highly successful and highly diverse. How, where and when human malaria parasites originated from within this diversity has long been a subject of fascination and sometimes also controversy. The past decade has seen the publication of a number of important discoveries about malaria parasite origins, all based on the application of molecular diagnostic tools to new sources of samples. This review summarizes some of those recent discoveries and discusses their implication for our current understanding of the origin and evolution of the Plasmodium genus. The nature of these discoveries and the manner in which they are made are then used to lay out a series of opportunities and challenges for the next wave of parasite hunters. -

Comparative Genomics of Ape Plasmodium Parasites Reveals Key Evolutionary Events Leading to Human Malaria
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations 2016 Comparative Genomics of Ape Plasmodium Parasites Reveals Key Evolutionary Events Leading to Human Malaria Sesh Alexander Sundararaman University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Evolution Commons, Genetics Commons, and the Microbiology Commons Recommended Citation Sundararaman, Sesh Alexander, "Comparative Genomics of Ape Plasmodium Parasites Reveals Key Evolutionary Events Leading to Human Malaria" (2016). Publicly Accessible Penn Dissertations. 2046. https://repository.upenn.edu/edissertations/2046 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/2046 For more information, please contact [email protected]. Comparative Genomics of Ape Plasmodium Parasites Reveals Key Evolutionary Events Leading to Human Malaria Abstract African great apes are infected with at least six species of P. falciparum-like parasites, including the ancestor of P. falciparum. Comparative studies of these parasites and P. falciparum (collectively termed the Laverania subgenus) will provide insight into the evolutionary origins of P. falciparum and identify genetic features that influence host tropism. Here we show that ape Laverania parasites do not serve as a recurrent source of human malaria and use novel enrichment techniques to derive near full-length genomes of close and distant relatives of P. falciparum. Using a combination of single template amplification and deep sequencing, we observe no evidence of ape Laverania infections in forest dwelling humans in Cameroon. This result supports previous findings that ape Laverania parasites are host specific and have successfully colonized humans only once. To understand the determinants of host specificity and identify genetic characteristics unique to P.