Debugging Parasite Genomes: Using Metabolic Modeling to Accelerate Antiparasitic Drug Development
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Exploration of Laboratory Techniques Relating to Cryptosporidium Parvum Propagation, Life Cycle Observation, and Host Immune Responses to Infection
EXPLORATION OF LABORATORY TECHNIQUES RELATING TO CRYPTOSPORIDIUM PARVUM PROPAGATION, LIFE CYCLE OBSERVATION, AND HOST IMMUNE RESPONSES TO INFECTION A Thesis Submitted to the Graduate Faculty of the North Dakota State University of Agriculture and Applied Science By Cheryl Marie Brown In Partial Fulfillment for the Degree of MASTER OF SCIENCE Major Department: Veterinary and Microbiological Sciences February 2014 Fargo, North Dakota North Dakota State University Graduate School Title EXPLORATION OF LABORATORY TECHNIQUES RELATING TO CRYPTOSPORIDIUM PARVUM PROPAGATION, LIFE CYCLE OBSERVATION, AND HOST IMMUNE RESPONSES TO INFECTION By Cheryl Marie Brown The Supervisory Committee certifies that this disquisition complies with North Dakota State University’s regulations and meets the accepted standards for the degree of MASTER OF SCIENCE SUPERVISORY COMMITTEE: Dr. Jane Schuh Chair Dr. John McEvoy Dr. Carrie Hammer Approved: 4-8-14 Dr. Charlene Wolf-Hall Date Department Chair ii ABSTRACT Cryptosporidium causes cryptosporidiosis, a self-limiting diarrheal disease in healthy people, but causes serious health issues for immunocompromised individuals. Cryptosporidiosis has been observed in humans since the early 1970s and continues to cause public health concerns. Cryptosporidium has a complicated life cycle making laboratory study challenging. This project explores several ways of studying Cryptosporidium parvum, with a goal of applying existing techniques to further understand this life cycle. Utilization of a neonatal mouse model demonstrated laser microdissection as a tool for studying host immune response to infeciton. A cell culture technique developed on FrameSlides™ enables laser microdissection of individual infected cells for further analysis. Finally, the hypothesis that the availability of cells to infect drives the switch from asexual to sexual parasite reproduction was tested by time-series infection. -

The Transcriptome of the Avian Malaria Parasite Plasmodium
bioRxiv preprint doi: https://doi.org/10.1101/072454; this version posted August 31, 2016. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 The Transcriptome of the Avian Malaria Parasite 2 Plasmodium ashfordi Displays Host-Specific Gene 3 Expression 4 5 6 7 8 Running title 9 The Transcriptome of Plasmodium ashfordi 10 11 Authors 12 Elin Videvall1, Charlie K. Cornwallis1, Dag Ahrén1,3, Vaidas Palinauskas2, Gediminas Valkiūnas2, 13 Olof Hellgren1 14 15 Affiliation 16 1Department of Biology, Lund University, Lund, Sweden 17 2Institute of Ecology, Nature Research Centre, Vilnius, Lithuania 18 3National Bioinformatics Infrastructure Sweden (NBIS), Lund University, Lund, Sweden 19 20 Corresponding authors 21 Elin Videvall ([email protected]) 22 Olof Hellgren ([email protected]) 23 24 1 bioRxiv preprint doi: https://doi.org/10.1101/072454; this version posted August 31, 2016. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 25 Abstract 26 27 Malaria parasites (Plasmodium spp.) include some of the world’s most widespread and virulent 28 pathogens, infecting a wide array of vertebrates. Our knowledge of the molecular mechanisms these 29 parasites use to invade and exploit hosts other than mice and primates is, however, extremely limited. 30 How do Plasmodium adapt to individual hosts and to the immune response of hosts throughout an 31 infection? To better understand parasite plasticity, and identify genes that are conserved across the 32 phylogeny, it is imperative that we characterize transcriptome-wide gene expression from non-model 33 malaria parasites in multiple host individuals. -

Plasmodium Falciparum Is Not As Lonely As Previously Considered
AUTOPHAGIC PUNCTUM ARTICLE ADDENDUM Virulence 2:1, 71-76; January/February 2011; © 2011 Landes Bioscience Plasmodium falciparum is not as lonely as previously considered Franck Prugnolle,1,* Francisco Ayala,2 Benjamin Ollomo,3 Céline Arnathau,1 Patrick Durand1 and François Renaud1,* 1Laboratoire MIVEGEC; UM1-CNRS 5290-IRD 224, IRD Montpellier, France; 2Department of Ecology and Evolutionary Biology; University of California; Irvine, CA USA; 3Centre International de Recherches Médicales de Franceville; Franceville, Gabon ntil very recently, only one species The identification of Plasmodium spe- U(P. reichenowi) was known to be a cies circulating in great apes in Africa phylogenetic sister lineage of P. falciparum, was primarily done during the first half the main malignant agent of human of the twentieth century, on the basis of malaria. In 2009 and 2010, new studies morphological features.1 This approach have revealed the existence of several new has several limitations.4 First, phenotypic phylogenetic species related to this deadly plasticity can lead to incorrect identifica- parasite and infecting chimpanzees and tions. Second, morphological keys are gorillas in Africa. These discoveries invite often effective only for a particular life us to explore a whole set of new questions, stage which cannot always be observed which we briefly do in this article. or is difficult to be. Finally, and perhaps most important, this approach overlooks The Plasmodium species infecting morphologically cryptic taxa. These limi- humans and non-human primates cluster tations, together with the difficulty to into two distinct phylogenetic lineages collect and manipulate great apes, were (Fig. 1). One of these lineages (in yellow certainly, at least in part, responsible for in Fig. -

Legionella Genus Genome Provide Multiple, Independent Combinations for Replication in Human Cells
Supplemental Material More than 18,000 effectors in the Legionella genus genome provide multiple, independent combinations for replication in human cells Laura Gomez-Valero1,2, Christophe Rusniok1,2, Danielle Carson3, Sonia Mondino1,2, Ana Elena Pérez-Cobas1,2, Monica Rolando1,2, Shivani Pasricha4, Sandra Reuter5+, Jasmin Demirtas1,2, Johannes Crumbach1,2, Stephane Descorps-Declere6, Elizabeth L. Hartland4,7,8, Sophie Jarraud9, Gordon Dougan5, Gunnar N. Schroeder3,10, Gad Frankel3, and Carmen Buchrieser1,2,* Table S1: Legionella strains analyzed in the present study Table S2: Type IV secretion systems predicted in the genomes analyzed Table S3: Eukaryotic like domains identified in the Legionella proteins analyzed Table S4: Small GTPases domains detected in the genus Legionella as defined in the CDD ncbi domain database Table S5: Eukaryotic like proteins detected in the Legionella genomes analyzed in this study Table S6: Aminoacid identity of the Dot/Icm components in Legionella species with respect to orthologous proteins in L. pneumophila Paris Table S7: Distribution of seventeen highly conserved Dot/Icm secreted substrates Table S8: Comparison of the effector reperotoire among strains of the same Legionella species Table S9. Number of Dot/Icm secreted proteins predicted in each strain analyzed Table S10: Replication capacity of the different Legionella species analyzed in this study and collection of literature data on Legionella replication Table S11: Orthologous table for all genes of the 80 analyzed strains based on PanOCT. The orthologoss where defined with the program PanOCT using the parameters previously indicated in material and methods.) Figure S1: Distribution of the genes predicted to encode for the biosynthesis of flagella among all Legionella species. -
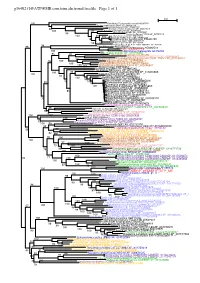
Page 1 G16482.T1@[email protected]
g16482.t1@[email protected] Page 1 of 1 0.2 100 Candidatus Thiomargarita nelsonii KHD07353 Bacillus thuringiensis Bt407 YP_006926419 Legionella pneumophila WP_014842957 88 Endozoicomonas montiporae WP_034879478 Acinetobacter baumannii WP_031968362 100 87 Vibrio nigripulchritudo WP_004402447 98 Psychrobacter sp. P11G3 WP_057760119 100 99 Xenorhabdus bovienii WP_038203911 100 Rouxiella chamberiensis WP_045046329 18 Dendroctonus ponderosae ERL83390 Serratia sp. M24T3 WP_009636677 98 56 100Lonsdalea quercina WP_026743324 Escherichia coli str. K-12 substr. MG1655 NP_415996 86 98 Chlorobi bacterium OLB5 KXK50071 CoptotermesTric_AAA92714 formosanus AGM32510 100 Histomonas meleagridis ACI16482 100Eucalyptus grandis XP_010033838 95 98 Ectocarpus siliculosus CBJ25596 Guillardia theta CCMP2712 XP_005838347 Phaeodactylum tricornutum CCAP 1055/1 XP_002180331 99 Aphanomyces astaci XP_009825348 95 100 Albugo laibachii Nc14 CCA15917 100 Phytophthora parasitica P1569 ETI54450 100 Phytophthora infestans T30-4 XP_002896607 100 Brugia malayi CDP93853 Bombus impatiens XP_003487345 Trichoplax adhaerens XP_002116081 95 Apteryx australis mantelli XP_013810665 100 100 Callorhinchus milii XP_007891153 Homo sapiens AAC50613 93 Callorhinchus milii XP_007900551 100 Clupea harengus XP_012674444 99 Chrysochloris asiatica XP_006865016 82 76 100 84Nannospalax galili XP_008842357 98 Cricetulus griseus XP_007626683 100 65Rousettus aegyptiacus XP_016015809 Condylura cristata XP_004683330 58Chinchilla lanigera XP_013367523 85 61Microcebus murinus XP_012594174 -

Paràsits Del Fílum Apicomplexa Presents a Larus Michahellis
Facultat de Ciències Memòria del Treball de Fi de Grau Paràsits del fílum Apicomplexa presents a Larus michahellis Laia Ruiz Ponsell Grau de Biologia Any acadèmic 2019-20 Treball tutelat per Claudia Paredes Esquivel Departament de Biologia S'autoritza la Universitat a incloure aquest treball en el Repositori Institucional Autor Tutor per a la seva consulta en accés obert i difusió en línia, amb finalitats Sí No Sí No exclusivament acadèmiques i d'investigació X X Paraules clau del treball: Toxoplasma gondii, toxoplasmosi, Larus michahellis, hoste, ELISA, ou, anticòs, antigen, infecció, Plasmodium, Haemoproteus, malària aviar, transmissió. Índex Resum ....................................................................................................................................... 1,2 Introducció ............................................................................................................................ 3-11 Generalitats de Larus michahellis ............................................................................................. 3 Generalitats del fílum Apicomplexa ...................................................................................... 4, 5 Cicle vital dels paràsits del fílum Apicomplexa .................................................................... 5, 6 Cicle vital de Toxoplasma gondii .......................................................................................... 6, 7 Epidemiologia i canvis comportamentals causats per T. gondii ............................................ 7, 8 -

Inferring Natural Selection Signals in Plasmodium Vivax-Encoded Proteins Having a Potential Role in Merozoite Invasion
Identificación de señales de selección natural en genes de Plasmodium vivax que codifican proteínas involucradas en el proceso de invasión para determinar su potencial uso en una vacuna antimalárica. Diego Edison Garzón Ospina Tesis Doctoral presentada como requisito para optar al título de Doctor en Ciencias Biomédicas y Biológicas de la Universidad del Rosario Bogotá, 2018 1 Identificación de señales de selección natural en genes de Plasmodium vivax que codifican proteínas involucradas en el proceso de invasión para determinar su potencial uso en una vacuna antimalárica. Estudiante Diego Edison Garzón Ospina Biólogo, Universidad INCCA de Colombia Magister en Ciencias-Microbiología, Universidad Nacional de Colombia Director Manuel Alfonso Patarroyo Gutiérrez M.D., Dr.Sc. Jefe del Departamento de Biología Molecular e Inmunología Fundación Instituto de Inmunología de Colombia (FIDIC) Profesor Titular, Escuela de Medicina y Ciencias de la Salud Universidad del Rosario DOCTORADO EN CIENCIAS BIOMÉDICAS Y BIOLÓGICAS UNIVERSIDAD DEL ROSARIO Bogotá, 2018 2 AGRADECIMIENTOS Quiero dar mis agradecimientos y dedicar este trabajo a mi madre: Martha Ospina Vargas, y a mis hermanos: Yamile Garzón Ospina y Julián David Escobar Ospina, quienes me han acompañado y apoyado en todo momento. A Sindy Paola Buitrago Puentes por su acompañamiento y apoyo durante este tiempo. También agradecer al Dr. Manuel Alfonso Patarroyo Gutiérrez, por haberme acogido en su equipo de trabajo y por apoyarme a lo largo de todos estos años. Quiero agradecer y reconocer la labor de: Andrea Estefanía Ramos, Darwin Andrés Moreno Pérez, Elizabeth Gutiérrez Vásquez, Heidy Daniela Ortiz Suarez, Lady Johanna Forero Rodríguez, Laura Alejandra Ricaurte Contreras, Leidy Paola Reyes, Luis Alfredo Baquero, Paola Andrea Camargo Ayala, Sindy Paola Buitrago Puentes, Ricardo De León Montero y Yimara Grosso Paz, quienes aportaron su tiempo, dedicación y esfuerzo, permitiendo la culminación de este trabajo. -

Catalogue of Protozoan Parasites Recorded in Australia Peter J. O
1 CATALOGUE OF PROTOZOAN PARASITES RECORDED IN AUSTRALIA PETER J. O’DONOGHUE & ROBERT D. ADLARD O’Donoghue, P.J. & Adlard, R.D. 2000 02 29: Catalogue of protozoan parasites recorded in Australia. Memoirs of the Queensland Museum 45(1):1-164. Brisbane. ISSN 0079-8835. Published reports of protozoan species from Australian animals have been compiled into a host- parasite checklist, a parasite-host checklist and a cross-referenced bibliography. Protozoa listed include parasites, commensals and symbionts but free-living species have been excluded. Over 590 protozoan species are listed including amoebae, flagellates, ciliates and ‘sporozoa’ (the latter comprising apicomplexans, microsporans, myxozoans, haplosporidians and paramyxeans). Organisms are recorded in association with some 520 hosts including mammals, marsupials, birds, reptiles, amphibians, fish and invertebrates. Information has been abstracted from over 1,270 scientific publications predating 1999 and all records include taxonomic authorities, synonyms, common names, sites of infection within hosts and geographic locations. Protozoa, parasite checklist, host checklist, bibliography, Australia. Peter J. O’Donoghue, Department of Microbiology and Parasitology, The University of Queensland, St Lucia 4072, Australia; Robert D. Adlard, Protozoa Section, Queensland Museum, PO Box 3300, South Brisbane 4101, Australia; 31 January 2000. CONTENTS the literature for reports relevant to contemporary studies. Such problems could be avoided if all previous HOST-PARASITE CHECKLIST 5 records were consolidated into a single database. Most Mammals 5 researchers currently avail themselves of various Reptiles 21 electronic database and abstracting services but none Amphibians 26 include literature published earlier than 1985 and not all Birds 34 journal titles are covered in their databases. Fish 44 Invertebrates 54 Several catalogues of parasites in Australian PARASITE-HOST CHECKLIST 63 hosts have previously been published. -

Plasmodium and Leucocytozoon (Sporozoa: Haemosporida) of Wild Birds in Bulgaria
Acta Protozool. (2003) 42: 205 - 214 Plasmodium and Leucocytozoon (Sporozoa: Haemosporida) of Wild Birds in Bulgaria Peter SHURULINKOV and Vassil GOLEMANSKY Institute of Zoology, Bulgarian Academy of Sciences, Sofia, Bulgaria Summary. Three species of parasites of the genus Plasmodium (P. relictum, P. vaughani, P. polare) and 6 species of the genus Leucocytozoon (L. fringillinarum, L. majoris, L. dubreuili, L. eurystomi, L. danilewskyi, L. bennetti) were found in the blood of 1332 wild birds of 95 species (mostly passerines), collected in the period 1997-2001. Data on the morphology, size, hosts, prevalence and infection intensity of the observed parasites are given. The total prevalence of the birds infected with Plasmodium was 6.2%. Plasmodium was observed in blood smears of 82 birds (26 species, all passerines). The highest prevalence of Plasmodium was found in the family Fringillidae: 18.5% (n=65). A high rate was also observed in Passeridae: 18.3% (n=71), Turdidae: 11.2% (n=98) and Paridae: 10.3% (n=68). The lowest prevalence was diagnosed in Hirundinidae: 2.5% (n=81). Plasmodium was found from March until October with no significant differences in the monthly values of the total prevalence. Resident birds were more often infected (13.2%, n=287) than locally nesting migratory birds (3.8%, n=213). Spring migrants and fall migrants were infected at almost the same rate of 4.2% (n=241) and 4.7% (n=529) respectively. Most infections were of low intensity (less than 1 parasite per 100 microscope fields at magnification 2000x). Leucocytozoon was found in 17 wild birds from 9 species (n=1332). -

(Haemosporida: Haemoproteidae), with Report of in Vitro Ookinetes of Haemoproteus Hirundi
Chagas et al. Parasites Vectors (2019) 12:422 https://doi.org/10.1186/s13071-019-3679-1 Parasites & Vectors RESEARCH Open Access Sporogony of four Haemoproteus species (Haemosporida: Haemoproteidae), with report of in vitro ookinetes of Haemoproteus hirundinis: phylogenetic inference indicates patterns of haemosporidian parasite ookinete development Carolina Romeiro Fernandes Chagas* , Dovilė Bukauskaitė, Mikas Ilgūnas, Rasa Bernotienė, Tatjana Iezhova and Gediminas Valkiūnas Abstract Background: Haemoproteus (Parahaemoproteus) species (Haemoproteidae) are widespread blood parasites that can cause disease in birds, but information about their vector species, sporogonic development and transmission remain fragmentary. This study aimed to investigate the complete sporogonic development of four Haemoproteus species in Culicoides nubeculosus and to test if phylogenies based on the cytochrome b gene (cytb) refect patterns of ookinete development in haemosporidian parasites. Additionally, one cytb lineage of Haemoproteus was identifed to the spe- cies level and the in vitro gametogenesis and ookinete development of Haemoproteus hirundinis was characterised. Methods: Laboratory-reared C. nubeculosus were exposed by allowing them to take blood meals on naturally infected birds harbouring single infections of Haemoproteus belopolskyi (cytb lineage hHIICT1), Haemoproteus hirun- dinis (hDELURB2), Haemoproteus nucleocondensus (hGRW01) and Haemoproteus lanii (hRB1). Infected insects were dissected at intervals in order to detect sporogonic stages. In vitro exfagellation, gametogenesis and ookinete development of H. hirundinis were also investigated. Microscopic examination and PCR-based methods were used to confrm species identity. Bayesian phylogenetic inference was applied to study the relationships among Haemopro- teus lineages. Results: All studied parasites completed sporogony in C. nubeculosus. Ookinetes and sporozoites were found and described. Development of H. hirundinis ookinetes was similar both in vivo and in vitro. -

Plasmodium Asexual Growth and Sexual Development in the Haematopoietic Niche of the Host
REVIEWS Plasmodium asexual growth and sexual development in the haematopoietic niche of the host Kannan Venugopal 1, Franziska Hentzschel1, Gediminas Valkiūnas2 and Matthias Marti 1* Abstract | Plasmodium spp. parasites are the causative agents of malaria in humans and animals, and they are exceptionally diverse in their morphology and life cycles. They grow and develop in a wide range of host environments, both within blood- feeding mosquitoes, their definitive hosts, and in vertebrates, which are intermediate hosts. This diversity is testament to their exceptional adaptability and poses a major challenge for developing effective strategies to reduce the disease burden and transmission. Following one asexual amplification cycle in the liver, parasites reach high burdens by rounds of asexual replication within red blood cells. A few of these blood- stage parasites make a developmental switch into the sexual stage (or gametocyte), which is essential for transmission. The bone marrow, in particular the haematopoietic niche (in rodents, also the spleen), is a major site of parasite growth and sexual development. This Review focuses on our current understanding of blood-stage parasite development and vascular and tissue sequestration, which is responsible for disease symptoms and complications, and when involving the bone marrow, provides a niche for asexual replication and gametocyte development. Understanding these processes provides an opportunity for novel therapies and interventions. Gametogenesis Malaria is one of the major life- threatening infectious Malaria parasites have a complex life cycle marked Maturation of male and female diseases in humans and is particularly prevalent in trop- by successive rounds of asexual replication across gametes. ical and subtropical low- income regions of the world. -

Malaria During the Last Decade1
MALARIA DURING THE LAST DECADE1 MARTIN D. YOUNG National Institutes of Health, National Microbiological Institute, Laboratory of Tropical Diseases, Columbia, South Carolina The starting point of this paper is rather arbitrarily set at January, 1942, but the selection of this date also has some significance in the knowledge of malaria. Much of the world had just become involved in a great war and was being con fronted with problems in disease control relative to the military. Of these diseases by far the most important was malaria. The following is concerned mainly with human malaria and is not intended to be a comprehensive review of the field but rather of those developments which appear to me to be significant. Of the tremendous amount of work that has gone on in the malaria of lower animals, reference will be made only to such as is particularly relevant to human malaria or that which can serve for comparison to point up the particular dis cussion at hand. BIOLOGY During this period little attention was paid to the cytology of the parasite- However, MacDougall (1947), studying Plasmodium vivax and P. falciparum and working specifically with gamete formation definitely established that chro mosomes were present in plasmodial parasites. Such had been indicated before but this was the first definitive proof. Additional work by Wolcott (unpublished) indicates that the asexual stages of P. vivax have two chromosomes. There have been no new species of human malaria parasites accepted during this period. Surveys and studies of infected military personnel have delineated more clearly the distribution of the recognized four species of malaria on a world wide basis and have shown that many strains, particularly of P.