Structural and Functional Insights Into Apicomplexan Gliding and Its Regulation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Non-Invasive Surveillance for Plasmodium in Reservoir Macaque
Siregar et al. Malar J (2015) 14:404 DOI 10.1186/s12936-015-0857-2 METHODOLOGY Open Access Non‑invasive surveillance for Plasmodium in reservoir macaque species Josephine E. Siregar1, Christina L. Faust2*, Lydia S. Murdiyarso1, Lis Rosmanah3, Uus Saepuloh3, Andrew P. Dobson2 and Diah Iskandriati3 Abstract Background: Primates are important reservoirs for human diseases, but their infection status and disease dynamics are difficult to track in the wild. Within the last decade, a macaque malaria, Plasmodium knowlesi, has caused disease in hundreds of humans in Southeast Asia. In order to track cases and understand zoonotic risk, it is imperative to be able to quantify infection status in reservoir macaque species. In this study, protocols for the collection of non-invasive samples and isolation of malaria parasites from naturally infected macaques are optimized. Methods: Paired faecal and blood samples from 60 Macaca fascicularis and four Macaca nemestrina were collected. All animals came from Sumatra or Java and were housed in semi-captive breeding colonies around West Java. DNA was extracted from samples using a modified protocol. Nested polymerase chain reactions (PCR) were run to detect Plasmodium using primers targeting mitochondrial DNA. Sensitivity of screening faecal samples for Plasmodium was compared to other studies using Kruskal Wallis tests and logistic regression models. Results: The best primer set was 96.7 % (95 % confidence intervals (CI): 83.3–99.4 %) sensitive for detecting Plasmo- dium in faecal samples of naturally infected macaques (n 30). This is the first study to produce definitive estimates of Plasmodium sensitivity and specificity in faecal samples= from naturally infected hosts. -

Plasmodium Vivax in Vitro Continuous Culture: the Spoke in the Wheel
Bermúdez et al. Malar J (2018) 17:301 https://doi.org/10.1186/s12936-018-2456-5 Malaria Journal REVIEW Open Access Plasmodium vivax in vitro continuous culture: the spoke in the wheel Maritza Bermúdez1, Darwin Andrés Moreno‑Pérez2,3, Gabriela Arévalo‑Pinzón1, Hernando Curtidor1,4 and Manuel Alfonso Patarroyo2,4* Abstract Understanding the life cycle of Plasmodium vivax is fundamental for developing strategies aimed at controlling and eliminating this parasitic species. Although advances in omic sciences and high-throughput techniques in recent years have enabled the identifcation and characterization of proteins which might be participating in P. vivax inva‑ sion of target cells, exclusive parasite tropism for invading reticulocytes has become the main obstacle in maintaining a continuous culture for this species. Such advance that would help in defning each parasite protein’s function in the complex process of P. vivax invasion, in addition to evaluating new therapeutic agents, is still a dream. Advances related to maintenance, culture medium supplements and the use of diferent sources of reticulocytes and parasites (strains and isolates) have been made regarding the development of an in vitro culture for P. vivax; however, only some cultures having few replication cycles have been obtained to date, meaning that this parasite’s maintenance goes beyond the technical components involved. Although it is still not yet clear which molecular mechanisms P. vivax prefers for invading young CD71+ reticulocytes [early maturation stages (I–II–III)], changes related to mem‑ brane proteins remodelling of such cells could form part of the explanation. The most relevant aspects regarding P. vivax in vitro culture and host cell characteristics have been analysed in this review to explain possible reasons why the species’ continuous in vitro culture is so difcult to standardize. -

University of Oklahoma
UNIVERSITY OF OKLAHOMA GRADUATE COLLEGE MACRONUTRIENTS SHAPE MICROBIAL COMMUNITIES, GENE EXPRESSION AND PROTEIN EVOLUTION A DISSERTATION SUBMITTED TO THE GRADUATE FACULTY in partial fulfillment of the requirements for the Degree of DOCTOR OF PHILOSOPHY By JOSHUA THOMAS COOPER Norman, Oklahoma 2017 MACRONUTRIENTS SHAPE MICROBIAL COMMUNITIES, GENE EXPRESSION AND PROTEIN EVOLUTION A DISSERTATION APPROVED FOR THE DEPARTMENT OF MICROBIOLOGY AND PLANT BIOLOGY BY ______________________________ Dr. Boris Wawrik, Chair ______________________________ Dr. J. Phil Gibson ______________________________ Dr. Anne K. Dunn ______________________________ Dr. John Paul Masly ______________________________ Dr. K. David Hambright ii © Copyright by JOSHUA THOMAS COOPER 2017 All Rights Reserved. iii Acknowledgments I would like to thank my two advisors Dr. Boris Wawrik and Dr. J. Phil Gibson for helping me become a better scientist and better educator. I would also like to thank my committee members Dr. Anne K. Dunn, Dr. K. David Hambright, and Dr. J.P. Masly for providing valuable inputs that lead me to carefully consider my research questions. I would also like to thank Dr. J.P. Masly for the opportunity to coauthor a book chapter on the speciation of diatoms. It is still such a privilege that you believed in me and my crazy diatom ideas to form a concise chapter in addition to learn your style of writing has been a benefit to my professional development. I’m also thankful for my first undergraduate research mentor, Dr. Miriam Steinitz-Kannan, now retired from Northern Kentucky University, who was the first to show the amazing wonders of pond scum. Who knew that studying diatoms and algae as an undergraduate would lead me all the way to a Ph.D. -

Plasmodium Falciparum Is Not As Lonely As Previously Considered
AUTOPHAGIC PUNCTUM ARTICLE ADDENDUM Virulence 2:1, 71-76; January/February 2011; © 2011 Landes Bioscience Plasmodium falciparum is not as lonely as previously considered Franck Prugnolle,1,* Francisco Ayala,2 Benjamin Ollomo,3 Céline Arnathau,1 Patrick Durand1 and François Renaud1,* 1Laboratoire MIVEGEC; UM1-CNRS 5290-IRD 224, IRD Montpellier, France; 2Department of Ecology and Evolutionary Biology; University of California; Irvine, CA USA; 3Centre International de Recherches Médicales de Franceville; Franceville, Gabon ntil very recently, only one species The identification of Plasmodium spe- U(P. reichenowi) was known to be a cies circulating in great apes in Africa phylogenetic sister lineage of P. falciparum, was primarily done during the first half the main malignant agent of human of the twentieth century, on the basis of malaria. In 2009 and 2010, new studies morphological features.1 This approach have revealed the existence of several new has several limitations.4 First, phenotypic phylogenetic species related to this deadly plasticity can lead to incorrect identifica- parasite and infecting chimpanzees and tions. Second, morphological keys are gorillas in Africa. These discoveries invite often effective only for a particular life us to explore a whole set of new questions, stage which cannot always be observed which we briefly do in this article. or is difficult to be. Finally, and perhaps most important, this approach overlooks The Plasmodium species infecting morphologically cryptic taxa. These limi- humans and non-human primates cluster tations, together with the difficulty to into two distinct phylogenetic lineages collect and manipulate great apes, were (Fig. 1). One of these lineages (in yellow certainly, at least in part, responsible for in Fig. -

Quantifying the Removal of Red Blood Cells in Macaca Mulatta During a Plasmodium Coatneyi Infection Luis L
Fonseca et al. Malar J (2016) 15:410 DOI 10.1186/s12936-016-1465-5 Malaria Journal RESEARCH Open Access Quantifying the removal of red blood cells in Macaca mulatta during a Plasmodium coatneyi infection Luis L. Fonseca1,4*, Harnel S. Alezi1, Alberto Moreno2,4, John W. Barnwell3,4, Mary R. Galinski2,4 and Eberhard O. Voit1,4 Abstract Background: Malaria is the most deadly parasitic disease in humans globally, and the long-time coexistence with malaria has left indelible marks in the human genome that are the causes of a variety of genetic disorders. Although anaemia is a common clinical complication of malaria, the root causes and mechanisms involved in the pathogenesis of malarial anaemia are unclear and difficult to study in humans. Non-human primate (NHP) model systems enable the mechanistic study and quantification of underlying causative factors of malarial anaemia, and particularly the onset of severe anaemia. Methods: Data were obtained in the course of Plasmodium coatneyi infections of malaria-naïve and semi-immune rhesus macaques (Macaca mulatta), whose red blood cells (RBCs) were labelled in situ with biotin at the time the infections were initiated. The data were used for a survival analysis that permitted, for the first time, an accurate esti- mation of the lifespan of erythrocytes in macaques. The data furthermore formed the basis for the development and parameterization of a recursive dynamic model of erythrocyte turnover, which was used for the quantification of RBC production and removal in each macaque. Results: The computational analysis demonstrated that the lifespan of erythrocytes in macaques is 98 21 days. -

Legionella Genus Genome Provide Multiple, Independent Combinations for Replication in Human Cells
Supplemental Material More than 18,000 effectors in the Legionella genus genome provide multiple, independent combinations for replication in human cells Laura Gomez-Valero1,2, Christophe Rusniok1,2, Danielle Carson3, Sonia Mondino1,2, Ana Elena Pérez-Cobas1,2, Monica Rolando1,2, Shivani Pasricha4, Sandra Reuter5+, Jasmin Demirtas1,2, Johannes Crumbach1,2, Stephane Descorps-Declere6, Elizabeth L. Hartland4,7,8, Sophie Jarraud9, Gordon Dougan5, Gunnar N. Schroeder3,10, Gad Frankel3, and Carmen Buchrieser1,2,* Table S1: Legionella strains analyzed in the present study Table S2: Type IV secretion systems predicted in the genomes analyzed Table S3: Eukaryotic like domains identified in the Legionella proteins analyzed Table S4: Small GTPases domains detected in the genus Legionella as defined in the CDD ncbi domain database Table S5: Eukaryotic like proteins detected in the Legionella genomes analyzed in this study Table S6: Aminoacid identity of the Dot/Icm components in Legionella species with respect to orthologous proteins in L. pneumophila Paris Table S7: Distribution of seventeen highly conserved Dot/Icm secreted substrates Table S8: Comparison of the effector reperotoire among strains of the same Legionella species Table S9. Number of Dot/Icm secreted proteins predicted in each strain analyzed Table S10: Replication capacity of the different Legionella species analyzed in this study and collection of literature data on Legionella replication Table S11: Orthologous table for all genes of the 80 analyzed strains based on PanOCT. The orthologoss where defined with the program PanOCT using the parameters previously indicated in material and methods.) Figure S1: Distribution of the genes predicted to encode for the biosynthesis of flagella among all Legionella species. -
Supplementary Figure 1 Multicenter Randomised Control Trial 2746 Randomised
Supplementary Figure 1 Multicenter randomised control trial 2746 randomised 947 control 910 MNP without zinc 889 MNP with zinc 223 lost to follow up 219 lost to follow up 183 lost to follow up 34 refused 29 refused 37 refused 16 died 12 died 9 died 3 excluded 4 excluded 1 excluded 671 in follow-up 646 in follow-up 659 in follow-up at 24mo of age at 24mo of age at 24mo of age Selection for Microbiome sequencing 516 paired samples unavailable 469 paired samples unavailable 497 paired samples unavailable 69 antibiotic use 63 antibiotic use 67 antibiotic use 31 outside of WLZ criteria 37 outside of WLZ criteria 34 outside of WLZ criteria 6 diarrhea last 7 days 2 diarrhea last 7 days 7 diarrhea last 7 days 39 WLZ > -1 at 24 mo 10 WLZ < -2 at 24mo 58 WLZ > -1 at 24 mo 17 WLZ < -2 at 24mo 48 WLZ > -1 at 24 mo 8 WLZ < -2 at 24mo available for selection available for selection available for selection available for selection available for selection1 available for selection1 14 selected 10 selected 15 selected 14 selected 20 selected1 7 selected1 1 Two subjects (one in the reference WLZ group and one undernourished) had, at 12 months, no diarrhea within 1 day of stool collection but reported diarrhea within 7 days prior. Length, cm kg Weight, Supplementary Figure 2. Length (left) and weight (right) z-scores of children recruited into clinical trial NCT00705445 during the first 24 months of life. Median and quantile values are shown, with medians for participants profiled in current study indicated by red (undernourished) and black (reference WLZ) lines. -

Why the –Omic Future of Apicomplexa Should Include Gregarines Julie Boisard, Isabelle Florent
Why the –omic future of Apicomplexa should include Gregarines Julie Boisard, Isabelle Florent To cite this version: Julie Boisard, Isabelle Florent. Why the –omic future of Apicomplexa should include Gregarines. Biology of the Cell, Wiley, 2020, 10.1111/boc.202000006. hal-02553206 HAL Id: hal-02553206 https://hal.archives-ouvertes.fr/hal-02553206 Submitted on 24 Apr 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Article title: Why the –omic future of Apicomplexa should include Gregarines. Names of authors: Julie BOISARD1,2 and Isabelle FLORENT1 Authors affiliations: 1. Molécules de Communication et Adaptation des Microorganismes (MCAM, UMR 7245), Département Adaptations du Vivant (AVIV), Muséum National d’Histoire Naturelle, CNRS, CP52, 57 rue Cuvier 75231 Paris Cedex 05, France. 2. Structure et instabilité des génomes (STRING UMR 7196 CNRS / INSERM U1154), Département Adaptations du vivant (AVIV), Muséum National d'Histoire Naturelle, CP 26, 57 rue Cuvier 75231 Paris Cedex 05, France. Short Title: Gregarines –omics for Apicomplexa studies -
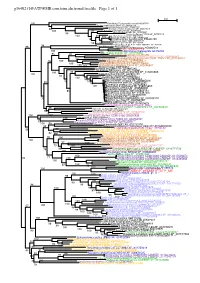
Page 1 G16482.T1@[email protected]
g16482.t1@[email protected] Page 1 of 1 0.2 100 Candidatus Thiomargarita nelsonii KHD07353 Bacillus thuringiensis Bt407 YP_006926419 Legionella pneumophila WP_014842957 88 Endozoicomonas montiporae WP_034879478 Acinetobacter baumannii WP_031968362 100 87 Vibrio nigripulchritudo WP_004402447 98 Psychrobacter sp. P11G3 WP_057760119 100 99 Xenorhabdus bovienii WP_038203911 100 Rouxiella chamberiensis WP_045046329 18 Dendroctonus ponderosae ERL83390 Serratia sp. M24T3 WP_009636677 98 56 100Lonsdalea quercina WP_026743324 Escherichia coli str. K-12 substr. MG1655 NP_415996 86 98 Chlorobi bacterium OLB5 KXK50071 CoptotermesTric_AAA92714 formosanus AGM32510 100 Histomonas meleagridis ACI16482 100Eucalyptus grandis XP_010033838 95 98 Ectocarpus siliculosus CBJ25596 Guillardia theta CCMP2712 XP_005838347 Phaeodactylum tricornutum CCAP 1055/1 XP_002180331 99 Aphanomyces astaci XP_009825348 95 100 Albugo laibachii Nc14 CCA15917 100 Phytophthora parasitica P1569 ETI54450 100 Phytophthora infestans T30-4 XP_002896607 100 Brugia malayi CDP93853 Bombus impatiens XP_003487345 Trichoplax adhaerens XP_002116081 95 Apteryx australis mantelli XP_013810665 100 100 Callorhinchus milii XP_007891153 Homo sapiens AAC50613 93 Callorhinchus milii XP_007900551 100 Clupea harengus XP_012674444 99 Chrysochloris asiatica XP_006865016 82 76 100 84Nannospalax galili XP_008842357 98 Cricetulus griseus XP_007626683 100 65Rousettus aegyptiacus XP_016015809 Condylura cristata XP_004683330 58Chinchilla lanigera XP_013367523 85 61Microcebus murinus XP_012594174 -

P. Falciparum, Replicates Within a Membrane-Bound Intraerythrocytic Parasitophorous Vacuole (PV)
Homing in on getting out: Characterisation of SERA6, a putative malarial protease with a role in egress Andrea Ruecker March 2012 MRC National Institute for Medical Research Division of Parasitology Mill Hill, London NW7 1AA Division of Infection and Immunity University College London This thesis is submitted to University College London for the degree of Doctor of Philosophy 1 Declaration Declaration I, Andrea Ruecker, confirm that the work presented in this thesis is my own. Where information has been derived from other sources, I confirm that this has been indicated in the thesis. Andrea Ruecker 2 Abstract Abstract The human malaria parasite, P. falciparum, replicates within a membrane-bound intraerythrocytic parasitophorous vacuole (PV). The resulting daughter merozoites actively escape from the host cell in a process called egress. There is convincing evidence that proteases are key players in this step. These proteases could serve as excellent targets for the development of new antimalarial drugs. P. falciparum Serine Repeat Antigens (SERAs) form a family of 9 proteins all containing a central papain- like domain that identifies them as putative cysteine proteases. They are highly conserved throughout all Plasmodium species, and there is strong genetic evidence that they may play a role in egress. P. falciparum SERA6 is one of the most highly- expressed SERAs in asexual erythrocyte stages. In this study biochemical fractionation and indirect immunofluorescence analysis were used to confirm localisation of SERA6 to the PV. It was shown that SERA6 is a substrate for PfSUB1, a subtilisin-like protease which is crucial for egress and which is released into the PV just prior to egress. -

A MOLECULAR PHYLOGENY of MALARIAL PARASITES RECOVERED from CYTOCHROME B GENE SEQUENCES
J. Parasitol., 88(5), 2002, pp. 972±978 q American Society of Parasitologists 2002 A MOLECULAR PHYLOGENY OF MALARIAL PARASITES RECOVERED FROM CYTOCHROME b GENE SEQUENCES Susan L. Perkins* and Jos. J. Schall Department of Biology, University of Vermont, Burlington, Vermont 05405. e-mail: [email protected] ABSTRACT: A phylogeny of haemosporidian parasites (phylum Apicomplexa, family Plasmodiidae) was recovered using mito- chondrial cytochrome b gene sequences from 52 species in 4 genera (Plasmodium, Hepatocystis, Haemoproteus, and Leucocy- tozoon), including parasite species infecting mammals, birds, and reptiles from over a wide geographic range. Leucocytozoon species emerged as an appropriate out-group for the other malarial parasites. Both parsimony and maximum-likelihood analyses produced similar phylogenetic trees. Life-history traits and parasite morphology, traditionally used as taxonomic characters, are largely phylogenetically uninformative. The Plasmodium and Hepatocystis species of mammalian hosts form 1 well-supported clade, and the Plasmodium and Haemoproteus species of birds and lizards form a second. Within this second clade, the relation- ships between taxa are more complex. Although jackknife support is weak, the Plasmodium of birds may form 1 clade and the Haemoproteus of birds another clade, but the parasites of lizards fall into several clusters, suggesting a more ancient and complex evolutionary history. The parasites currently placed within the genus Haemoproteus may not be monophyletic. Plasmodium falciparum of humans was not derived from an avian malarial ancestor and, except for its close sister species, P. reichenowi,is only distantly related to haemospordian parasites of all other mammals. Plasmodium is paraphyletic with respect to 2 other genera of malarial parasites, Haemoproteus and Hepatocystis. -

Inferring Natural Selection Signals in Plasmodium Vivax-Encoded Proteins Having a Potential Role in Merozoite Invasion
Identificación de señales de selección natural en genes de Plasmodium vivax que codifican proteínas involucradas en el proceso de invasión para determinar su potencial uso en una vacuna antimalárica. Diego Edison Garzón Ospina Tesis Doctoral presentada como requisito para optar al título de Doctor en Ciencias Biomédicas y Biológicas de la Universidad del Rosario Bogotá, 2018 1 Identificación de señales de selección natural en genes de Plasmodium vivax que codifican proteínas involucradas en el proceso de invasión para determinar su potencial uso en una vacuna antimalárica. Estudiante Diego Edison Garzón Ospina Biólogo, Universidad INCCA de Colombia Magister en Ciencias-Microbiología, Universidad Nacional de Colombia Director Manuel Alfonso Patarroyo Gutiérrez M.D., Dr.Sc. Jefe del Departamento de Biología Molecular e Inmunología Fundación Instituto de Inmunología de Colombia (FIDIC) Profesor Titular, Escuela de Medicina y Ciencias de la Salud Universidad del Rosario DOCTORADO EN CIENCIAS BIOMÉDICAS Y BIOLÓGICAS UNIVERSIDAD DEL ROSARIO Bogotá, 2018 2 AGRADECIMIENTOS Quiero dar mis agradecimientos y dedicar este trabajo a mi madre: Martha Ospina Vargas, y a mis hermanos: Yamile Garzón Ospina y Julián David Escobar Ospina, quienes me han acompañado y apoyado en todo momento. A Sindy Paola Buitrago Puentes por su acompañamiento y apoyo durante este tiempo. También agradecer al Dr. Manuel Alfonso Patarroyo Gutiérrez, por haberme acogido en su equipo de trabajo y por apoyarme a lo largo de todos estos años. Quiero agradecer y reconocer la labor de: Andrea Estefanía Ramos, Darwin Andrés Moreno Pérez, Elizabeth Gutiérrez Vásquez, Heidy Daniela Ortiz Suarez, Lady Johanna Forero Rodríguez, Laura Alejandra Ricaurte Contreras, Leidy Paola Reyes, Luis Alfredo Baquero, Paola Andrea Camargo Ayala, Sindy Paola Buitrago Puentes, Ricardo De León Montero y Yimara Grosso Paz, quienes aportaron su tiempo, dedicación y esfuerzo, permitiendo la culminación de este trabajo.