This Thesis Has Been Submitted in Fulfilment of the Requirements for a Postgraduate Degree (E.G
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

~.. R---'------ : KASMERA: Vol
~.. r---'-------------- : KASMERA: Vol.. 9, No. 1 4,1981 Zulla. Maracaibo. Venezuela. PROTOZOOS DE VENEZUELA Carlos Diaz Ungrla· Tratamos con este trabajo de ofrecer una puesta al día de los protozoos estudiados en nuestro país. Con ello damos un anticipo de lo que será nuestra próxima obra, en la cual, además de actualizar los problemas taxonómicos, pensamos hacer énfasis en la ultraestructura, cuyo cono cimiento es básico hoy día para manejar los protozoos, comQ animales unicelulares que son. Igualmente tratamos de difundir en nuestro medio la clasificación ac tual, que difiere tanto de la que se sigue estudiando. y por último, tratamos de reunir en un solo trabajo toda la infor mación bibliográfica venezolana, ya que es sabido que nuestros autores se ven precisados a publicar en revistas foráneas, y esto se ha acentuado en los últimos diez (10) años. En nuestro trabajo presentaremos primero la lista alfabética de los protozoos venezolanos, después ofreceremos su clasificación, para terminar por distribuirlos de acuerdo a sus hospedadores . • Profesor de la Facultad de Ciencias Veterinarias de la Universidad del Zulia. Maracaibo-Venezuela. -147 Con la esperanza de que nuestro trabajo sea útil anuestros colegas. En Maracaibo, abril de mil novecientos ochenta. 1 LISTA ALF ABETICA DE LOS PROTOZOOS DE VENEZUELA Babesia (Babesia) bigemina, Smith y Kilbome, 1893. Seflalada en Bos taurus por Zieman (1902). Deutsch. Med. Wochens., 20 y 21. Babesia (Babesia) caballi Nuttall y Stricldand. 1910. En Equus cabal/uso Gallo y Vogelsang (1051). Rev. Med.Vet. y Par~. 10 (1-4); 3. Babesia (Babesia) canis. Piana y Galli Valerio, 1895. En Canis ¡ami/iaris. -

Plasmodium Scientific Classification
Plasmodium - Wikipedia https://en.wikipedia.org/wiki/Plasmodium From Wikipedia, the free encyclopedia Plasmodium is a genus of parasitic alveolates, many of which cause malaria in their hosts.[1] The parasite always has two hosts in its life Plasmodium cycle: a Dipteran insect host and a vertebrate host. Sexual reproduction always occurs in the insect, making it the definitive host.[2] The life-cycles of Plasmodium species involve several different stages both in the insect and the vertebrate host. These stages include sporozoites, which are injected by the insect vector into the vertebrate host's blood. Sporozoites infect the host liver, giving rise to merozoites and (in some species) hypnozoites. These move into the blood where they infect red blood cells. In the red blood cells, the parasites can either form more merozoites to infect more red blood cells, or produce gametocytes which are taken up by insects which feed on the vertebrate host. In the insect host, gametocytes merge to sexually reproduce. After sexual reproduction, parasites grow into new sporozoites, which move to the insect's salivary glands, from which they can infect a vertebrate False-colored electron micrograph of a [1] host bitten by the insect. Plasmodium sp. sporozoite. The genus Plasmodium was first described in 1885. It now contains Scientific classification about 200 species, which are spread across the world where both the (unranked): SAR insect and vertebrate hosts are present. Five species regularly infect humans, while many others infect birds, reptiles, -

Transcriptomic Analysis Reveals Evidence for a Cryptic Plastid in the Colpodellid Voromonas Pontica, a Close Relative of Chromerids and Apicomplexan Parasites
Transcriptomic Analysis Reveals Evidence for a Cryptic Plastid in the Colpodellid Voromonas pontica, a Close Relative of Chromerids and Apicomplexan Parasites Gillian H. Gile*, Claudio H. Slamovits Department of Biochemistry and Molecular Biology, Dalhousie University, Halifax, Nova Scotia, Canada Abstract Colpodellids are free-living, predatory flagellates, but their close relationship to photosynthetic chromerids and plastid- bearing apicomplexan parasites suggests they were ancestrally photosynthetic. Colpodellids may therefore retain a cryptic plastid, or they may have lost their plastids entirely, like the apicomplexan Cryptosporidium. To find out, we generated transcriptomic data from Voromonas pontica ATCC 50640 and searched for homologs of genes encoding proteins known to function in the apicoplast, the non-photosynthetic plastid of apicomplexans. We found candidate genes from multiple plastid-associated pathways including iron-sulfur cluster assembly, isoprenoid biosynthesis, and tetrapyrrole biosynthesis, along with a plastid-type phosphate transporter gene. Four of these sequences include the 59 end of the coding region and are predicted to encode a signal peptide and a transit peptide-like region. This is highly suggestive of targeting to a cryptic plastid. We also performed a taxon-rich phylogenetic analysis of small subunit ribosomal RNA sequences from colpodellids and their relatives, which suggests that photosynthesis was lost more than once in colpodellids, and independently in V. pontica and apicomplexans. Colpodellids therefore represent a valuable source of comparative data for understanding the process of plastid reduction in humanity’s most deadly parasite. Citation: Gile GH, Slamovits CH (2014) Transcriptomic Analysis Reveals Evidence for a Cryptic Plastid in the Colpodellid Voromonas pontica, a Close Relative of Chromerids and Apicomplexan Parasites. -

Plasmodium Falciparum Is Not As Lonely As Previously Considered
AUTOPHAGIC PUNCTUM ARTICLE ADDENDUM Virulence 2:1, 71-76; January/February 2011; © 2011 Landes Bioscience Plasmodium falciparum is not as lonely as previously considered Franck Prugnolle,1,* Francisco Ayala,2 Benjamin Ollomo,3 Céline Arnathau,1 Patrick Durand1 and François Renaud1,* 1Laboratoire MIVEGEC; UM1-CNRS 5290-IRD 224, IRD Montpellier, France; 2Department of Ecology and Evolutionary Biology; University of California; Irvine, CA USA; 3Centre International de Recherches Médicales de Franceville; Franceville, Gabon ntil very recently, only one species The identification of Plasmodium spe- U(P. reichenowi) was known to be a cies circulating in great apes in Africa phylogenetic sister lineage of P. falciparum, was primarily done during the first half the main malignant agent of human of the twentieth century, on the basis of malaria. In 2009 and 2010, new studies morphological features.1 This approach have revealed the existence of several new has several limitations.4 First, phenotypic phylogenetic species related to this deadly plasticity can lead to incorrect identifica- parasite and infecting chimpanzees and tions. Second, morphological keys are gorillas in Africa. These discoveries invite often effective only for a particular life us to explore a whole set of new questions, stage which cannot always be observed which we briefly do in this article. or is difficult to be. Finally, and perhaps most important, this approach overlooks The Plasmodium species infecting morphologically cryptic taxa. These limi- humans and non-human primates cluster tations, together with the difficulty to into two distinct phylogenetic lineages collect and manipulate great apes, were (Fig. 1). One of these lineages (in yellow certainly, at least in part, responsible for in Fig. -

Legionella Genus Genome Provide Multiple, Independent Combinations for Replication in Human Cells
Supplemental Material More than 18,000 effectors in the Legionella genus genome provide multiple, independent combinations for replication in human cells Laura Gomez-Valero1,2, Christophe Rusniok1,2, Danielle Carson3, Sonia Mondino1,2, Ana Elena Pérez-Cobas1,2, Monica Rolando1,2, Shivani Pasricha4, Sandra Reuter5+, Jasmin Demirtas1,2, Johannes Crumbach1,2, Stephane Descorps-Declere6, Elizabeth L. Hartland4,7,8, Sophie Jarraud9, Gordon Dougan5, Gunnar N. Schroeder3,10, Gad Frankel3, and Carmen Buchrieser1,2,* Table S1: Legionella strains analyzed in the present study Table S2: Type IV secretion systems predicted in the genomes analyzed Table S3: Eukaryotic like domains identified in the Legionella proteins analyzed Table S4: Small GTPases domains detected in the genus Legionella as defined in the CDD ncbi domain database Table S5: Eukaryotic like proteins detected in the Legionella genomes analyzed in this study Table S6: Aminoacid identity of the Dot/Icm components in Legionella species with respect to orthologous proteins in L. pneumophila Paris Table S7: Distribution of seventeen highly conserved Dot/Icm secreted substrates Table S8: Comparison of the effector reperotoire among strains of the same Legionella species Table S9. Number of Dot/Icm secreted proteins predicted in each strain analyzed Table S10: Replication capacity of the different Legionella species analyzed in this study and collection of literature data on Legionella replication Table S11: Orthologous table for all genes of the 80 analyzed strains based on PanOCT. The orthologoss where defined with the program PanOCT using the parameters previously indicated in material and methods.) Figure S1: Distribution of the genes predicted to encode for the biosynthesis of flagella among all Legionella species. -

The Nuclear 18S Ribosomal Dnas of Avian Haemosporidian Parasites Josef Harl1, Tanja Himmel1, Gediminas Valkiūnas2 and Herbert Weissenböck1*
Harl et al. Malar J (2019) 18:305 https://doi.org/10.1186/s12936-019-2940-6 Malaria Journal RESEARCH Open Access The nuclear 18S ribosomal DNAs of avian haemosporidian parasites Josef Harl1, Tanja Himmel1, Gediminas Valkiūnas2 and Herbert Weissenböck1* Abstract Background: Plasmodium species feature only four to eight nuclear ribosomal units on diferent chromosomes, which are assumed to evolve independently according to a birth-and-death model, in which new variants origi- nate by duplication and others are deleted throughout time. Moreover, distinct ribosomal units were shown to be expressed during diferent developmental stages in the vertebrate and mosquito hosts. Here, the 18S rDNA sequences of 32 species of avian haemosporidian parasites are reported and compared to those of simian and rodent Plasmodium species. Methods: Almost the entire 18S rDNAs of avian haemosporidians belonging to the genera Plasmodium (7), Haemo- proteus (9), and Leucocytozoon (16) were obtained by PCR, molecular cloning, and sequencing ten clones each. Phy- logenetic trees were calculated and sequence patterns were analysed and compared to those of simian and rodent malaria species. A section of the mitochondrial CytB was also sequenced. Results: Sequence patterns in most avian Plasmodium species were similar to those in the mammalian parasites with most species featuring two distinct 18S rDNA sequence clusters. Distinct 18S variants were also found in Haemopro- teus tartakovskyi and the three Leucocytozoon species, whereas the other species featured sets of similar haplotypes. The 18S rDNA GC-contents of the Leucocytozoon toddi complex and the subgenus Parahaemoproteus were extremely high with 49.3% and 44.9%, respectively. -
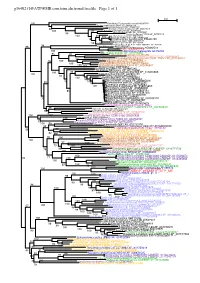
Page 1 G16482.T1@[email protected]
g16482.t1@[email protected] Page 1 of 1 0.2 100 Candidatus Thiomargarita nelsonii KHD07353 Bacillus thuringiensis Bt407 YP_006926419 Legionella pneumophila WP_014842957 88 Endozoicomonas montiporae WP_034879478 Acinetobacter baumannii WP_031968362 100 87 Vibrio nigripulchritudo WP_004402447 98 Psychrobacter sp. P11G3 WP_057760119 100 99 Xenorhabdus bovienii WP_038203911 100 Rouxiella chamberiensis WP_045046329 18 Dendroctonus ponderosae ERL83390 Serratia sp. M24T3 WP_009636677 98 56 100Lonsdalea quercina WP_026743324 Escherichia coli str. K-12 substr. MG1655 NP_415996 86 98 Chlorobi bacterium OLB5 KXK50071 CoptotermesTric_AAA92714 formosanus AGM32510 100 Histomonas meleagridis ACI16482 100Eucalyptus grandis XP_010033838 95 98 Ectocarpus siliculosus CBJ25596 Guillardia theta CCMP2712 XP_005838347 Phaeodactylum tricornutum CCAP 1055/1 XP_002180331 99 Aphanomyces astaci XP_009825348 95 100 Albugo laibachii Nc14 CCA15917 100 Phytophthora parasitica P1569 ETI54450 100 Phytophthora infestans T30-4 XP_002896607 100 Brugia malayi CDP93853 Bombus impatiens XP_003487345 Trichoplax adhaerens XP_002116081 95 Apteryx australis mantelli XP_013810665 100 100 Callorhinchus milii XP_007891153 Homo sapiens AAC50613 93 Callorhinchus milii XP_007900551 100 Clupea harengus XP_012674444 99 Chrysochloris asiatica XP_006865016 82 76 100 84Nannospalax galili XP_008842357 98 Cricetulus griseus XP_007626683 100 65Rousettus aegyptiacus XP_016015809 Condylura cristata XP_004683330 58Chinchilla lanigera XP_013367523 85 61Microcebus murinus XP_012594174 -

The Revised Classification of Eukaryotes
See discussions, stats, and author profiles for this publication at: https://www.researchgate.net/publication/231610049 The Revised Classification of Eukaryotes Article in Journal of Eukaryotic Microbiology · September 2012 DOI: 10.1111/j.1550-7408.2012.00644.x · Source: PubMed CITATIONS READS 961 2,825 25 authors, including: Sina M Adl Alastair Simpson University of Saskatchewan Dalhousie University 118 PUBLICATIONS 8,522 CITATIONS 264 PUBLICATIONS 10,739 CITATIONS SEE PROFILE SEE PROFILE Christopher E Lane David Bass University of Rhode Island Natural History Museum, London 82 PUBLICATIONS 6,233 CITATIONS 464 PUBLICATIONS 7,765 CITATIONS SEE PROFILE SEE PROFILE Some of the authors of this publication are also working on these related projects: Biodiversity and ecology of soil taste amoeba View project Predator control of diversity View project All content following this page was uploaded by Smirnov Alexey on 25 October 2017. The user has requested enhancement of the downloaded file. The Journal of Published by the International Society of Eukaryotic Microbiology Protistologists J. Eukaryot. Microbiol., 59(5), 2012 pp. 429–493 © 2012 The Author(s) Journal of Eukaryotic Microbiology © 2012 International Society of Protistologists DOI: 10.1111/j.1550-7408.2012.00644.x The Revised Classification of Eukaryotes SINA M. ADL,a,b ALASTAIR G. B. SIMPSON,b CHRISTOPHER E. LANE,c JULIUS LUKESˇ,d DAVID BASS,e SAMUEL S. BOWSER,f MATTHEW W. BROWN,g FABIEN BURKI,h MICAH DUNTHORN,i VLADIMIR HAMPL,j AARON HEISS,b MONA HOPPENRATH,k ENRIQUE LARA,l LINE LE GALL,m DENIS H. LYNN,n,1 HILARY MCMANUS,o EDWARD A. D. -

A Meta-Analysis of the Genus Alouatta
Chapter 17 Ecological and Anthropogenic Influences on Patterns of Parasitism in Free-Ranging Primates: A Meta-analysis of the Genus Alouatta Martin M. Kowalewski and Thomas R. Gillespie 17.1 Introduction Parasites play a central role in tropical ecosystems, affecting the ecology and evolution of species interactions, host population growth and regulation, and com- munity biodiversity (Esch and Fernandez 1993; Hudson, Dobson and Newborn 1998; Hochachka and Dhondt 2000; Hudson et al. 2002). Our understanding of how nat- ural and anthropogenic factors affect host-parasite dynamics in free-ranging pri- mate populations (Gillespie, Chapman and Greiner 2005a; Gillespie, Greiner and Chapman 2005b; Gillespie and Chapman 2006) and the relationship between wild primates and human health in rural or remote areas (McGrew et al. 1989; Stuart et al. 1990; Muller-Graf, Collins and Woolhouse 1997; Gillespie et al. 2005b; Pedersen et al. 2005) remain largely unexplored. The majority of emerging infec- tious diseases are zoonotic – easily transferred among humans, wildlife, and domes- ticated animals – (Nunn and Altizer 2006). For example, Taylor, Latham and Woolhouse (2001) found that 61% of human pathogens are shared with animal hosts. Identifying general principles governing parasite occurrence and prevalence is critical for planning animal conservation and protecting human health (Nunn et al. 2003). In this review, we examine how various ecological and anthropogenic factors affect patterns of parasitism in free-ranging howler monkeys (Genus Alouatta). 17.1.1 Evidence of the Relationships Between Howlers and Parasitic Diseases in South America The genus Alouatta is the most geographically widespread non-human primate in South America, with 8 of 10 Alouatta species ranging from Northern Colombia M.M. -

Таксономический Ранг И Место В Системе Протистов Colpodellida1
ПАРАЗИТОЛОГИЯ, 34, 7, 2000 УДК 576.893.19+ 593.19 ТАКСОНОМИЧЕСКИЙ РАНГ И МЕСТО В СИСТЕМЕ ПРОТИСТОВ COLPODELLIDA1 © А. П. Мыльников, М. В. Крылов, А. О. Фролов Анализ морфофункциональной организации и дивергентных процессов у Colpodellida, Perkinsida, Gregrinea, Coccidea подтвердил наличие у них уникального общего плана строения и необходимость объединения в один тип Sporozoa. Таксономический ранг и место в системе Colpodellida представляется следующим образом: тип Sporozoa Leuckart 1879; em. Krylov, Mylnikov, 1986 (Syn.: Apicomplexa Levine, 1970). Хищники либо паразиты. Имеют общий план строения: пелликулу, состоящую у расселительных стадий из плазматической мембраны и внутреннего мембранного комплекса, микропору(ы), субпелликулярные микротрубочки, коноид (у части редуцирован), роптрии и микронемы (у части редуцированы), трубчатые кристы в митохондриях. Класс Perkinsea Levine, 1978. Хищники или паразиты, имеющие в жизненном цикле вегетативные двужгутиковые стадии развития. Подкласс 1. Colpodellia nom. nov. (Syn.: Spiromonadia Krylov, Mylnikov, 1986). Хищники; имеют два гетеродинамных жгутика; масти- геномы нитевидные (если имеются); цисты 2—4-ядерные; стрекательные органеллы — трихо- цисты. Подкласс 2. Perkinsia Levine, 1978. Все виды — паразиты; зооспоры имеют два гетеродинамных жгутика; мастигонемы (если имеются) нитевидные и в виде щетинок. Главная цель систематиков — построение естественной системы. Естественная система прежде всего должна обладать максимальными прогностическими свойствами (Старобогатов, 1989). Иными словами, знания -

Ungulate Malaria Parasites Thomas J
www.nature.com/scientificreports OPEN Ungulate malaria parasites Thomas J. Templeton1,2,*, Masahito Asada1,*, Montakan Jiratanh3, Sohta A. Ishikawa4,5, Sonthaya Tiawsirisup6, Thillaiampalam Sivakumar7, Boniface Namangala8, Mika Takeda1, Kingdao Mohkaew3, Supawan Ngamjituea3, Noboru Inoue7, Chihiro Sugimoto9, Yuji Inagaki5,10, Yasuhiko Suzuki9, Naoaki Yokoyama7, Morakot Kaewthamasorn11 & Osamu Kaneko1 Received: 14 December 2015 Accepted: 02 March 2016 Haemosporida parasites of even-toed ungulates are diverse and globally distributed, but since their discovery in 1913 their characterization has relied exclusively on microscopy-based descriptions. In Published: 21 March 2016 order to bring molecular approaches to bear on the identity and evolutionary relationships of ungulate malaria parasites, we conducted Plasmodium cytb-specific nested PCR surveys using blood from water buffalo in Vietnam and Thailand, and goats in Zambia. We found thatPlasmodium is readily detectable from water buffalo in these countries, indicating that buffaloPlasmodium is distributed in a wider region than India, which is the only area in which buffaloPlasmodium has been reported. Two types (I and II) of Plasmodium sequences were identified from water buffalo and a third type (III) was isolated from goat. Morphology of the parasite was confirmed in Giemsa-reagent stained blood smears for the Type I sample. Complete mitochondrial DNA sequences were isolated and used to infer a phylogeny in which ungulate malaria parasites form a monophyletic clade within the Haemosporida, and branch prior to the clade containing bird, lizard and other mammalian Plasmodium. Thus it is likely that host switching of Plasmodium from birds to mammals occurred multiple times, with a switch to ungulates independently from other mammalian Plasmodium. -

Complex Communities of Small Protists and Unexpected Occurrence Of
Complex communities of small protists and unexpected occurrence of typical marine lineages in shallow freshwater systems Marianne Simon, Ludwig Jardillier, Philippe Deschamps, David Moreira, Gwendal Restoux, Paola Bertolino, Purificación López-García To cite this version: Marianne Simon, Ludwig Jardillier, Philippe Deschamps, David Moreira, Gwendal Restoux, et al.. Complex communities of small protists and unexpected occurrence of typical marine lineages in shal- low freshwater systems. Environmental Microbiology, Society for Applied Microbiology and Wiley- Blackwell, 2015, 17 (10), pp.3610-3627. 10.1111/1462-2920.12591. hal-03022575 HAL Id: hal-03022575 https://hal.archives-ouvertes.fr/hal-03022575 Submitted on 24 Nov 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Europe PMC Funders Group Author Manuscript Environ Microbiol. Author manuscript; available in PMC 2015 October 26. Published in final edited form as: Environ Microbiol. 2015 October ; 17(10): 3610–3627. doi:10.1111/1462-2920.12591. Europe PMC Funders Author Manuscripts Complex communities of small protists and unexpected occurrence of typical marine lineages in shallow freshwater systems Marianne Simon, Ludwig Jardillier, Philippe Deschamps, David Moreira, Gwendal Restoux, Paola Bertolino, and Purificación López-García* Unité d’Ecologie, Systématique et Evolution, CNRS UMR 8079, Université Paris-Sud, 91405 Orsay, France Summary Although inland water bodies are more heterogeneous and sensitive to environmental variation than oceans, the diversity of small protists in these ecosystems is much less well-known.