Activation of Ras Requires the ERM-Dependent Link of Actin to the Plasma Membrane
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
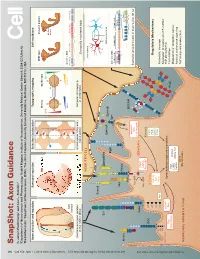
Snapshot: Axon Guidance Pasterkamp R
494 1 Cell Cell ??? SnapShot: Axon Guidance 153 SnapShot: XXXXXXXXXXXXXXXXXXXXXXXXXX 1 2 , ??MONTH?? ??DATE??, 200? ©200? Elsevier Inc. 200?©200? ElsevierInc. , ??MONTH?? ??DATE??, DOI R. Jeroen Pasterkamp and Alex L. Kolodkin , April11, 2013©2013Elsevier Inc. DOI http://dx.doi.org/10.1016/j.cell.2013.03.031 AUTHOR XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX 1 AFFILIATIONDepartment of XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX Neuroscience and Pharmacology, Rudolf Magnus Institute of Neuroscience, University Medical Center Utrecht, 3584 CG Utrecht, The Netherlands; 2Department of Neuroscience, HHMI, The Johns Hopkins University School of Medicine, Baltimore, MD 21212, USA Axon attraction and repulsion Surround repulsion Selective fasciculation Topographic mapping Self-avoidance Wild-type Dscam1 mutant Sema3A A Retina SC/Tectum EphB EphrinB P D D Mutant T A neuron N P A V V Genomic DNA P EphA EphrinA Slit Exon 4 (12) Exon 6 (48) Exon 9 (33) Exon 17 (2) Netrin Commissural axon guidance Surround repulsion of peripheral Grasshopper CNS axon Retinotectal mapping at the CNS midline nerves in vertebrates fasciculation in vertebrates Drosophila mushroom body XXXXXXXXX NEURITE/CELL Isoneuronal Sema3 Slit Sema1/4-6 Heteroneuronal EphrinA EphrinB FasII Eph Genomic DNA Pcdh-α (14) Pcdh-β (22) Pcdh-γ (22) Variable Con Variable Con Nrp Plexin ** *** Netrin LAMELLIPODIA Con DSCAM ephexin Starburst amacrine cells in mammalian retina Ras-GTP Vav See online version for legend and references. α-chimaerin GEFs/GAPs Robo FARP Ras-GDP LARG RhoGEF Kinases DCC cc0 GTPases PKA cc1 FAK Regulatory Mechanisms See online versionfor??????. Cdc42 GSK3 cc2 Rac PI3K P1 Rho P2 cc3 Abl Proteolytic cleavage P3 Regulation of expression (TF, miRNA, FILOPODIA srGAP Cytoskeleton regulatory proteins multiple isoforms) Sos Trio Pcdh Cis inhibition DOCK180 PAK ROCK Modulation of receptors’ output LIMK Myosin-II Colin Forward and reverse signaling Actin Trafcking and endocytosis NEURONAL GROWTH CONE Microtubules SnapShot: Axon Guidance R. -

Adaptive Stress Signaling in Targeted Cancer Therapy Resistance
Oncogene (2015) 34, 5599–5606 © 2015 Macmillan Publishers Limited All rights reserved 0950-9232/15 www.nature.com/onc REVIEW Adaptive stress signaling in targeted cancer therapy resistance E Pazarentzos1,2 and TG Bivona1,2 The identification of specific genetic alterations that drive the initiation and progression of cancer and the development of targeted drugs that act against these driver alterations has revolutionized the treatment of many human cancers. Although substantial progress has been achieved with the use of such targeted cancer therapies, resistance remains a major challenge that limits the overall clinical impact. Hence, despite progress, new strategies are needed to enhance response and eliminate resistance to targeted cancer therapies in order to achieve durable or curative responses in patients. To date, efforts to characterize mechanisms of resistance have primarily focused on molecular events that mediate primary or secondary resistance in patients. Less is known about the initial molecular response and adaptation that may occur in tumor cells early upon exposure to a targeted agent. Although understudied, emerging evidence indicates that the early adaptive changes by which tumor cells respond to the stress of a targeted therapy may be crucial for tumo r cell survival during treatment and the development of resistance. Here we review recent data illuminating the molecular architecture underlying adaptive stress signaling in tumor cells. We highlight how leveraging this knowledge could catalyze novel strategies to minimize -

Regulation of the Mammalian Target of Rapamycin Complex 2 (Mtorc2)
Regulation of the Mammalian Target Of Rapamycin Complex 2 (mTORC2) Inauguraldissertation Zur Erlangung der Würde eines Doktors der Philosophie vorgelegt der Philosophisch-Naturwissenschaftlichen Fakultät der Universität Basel von Klaus-Dieter Molle aus Heilbronn, Deutschland Basel, 2006 Genehmigt von der Philosophisch-Naturwissenschaftlichen Fakultät Auf Antrag von Prof. Michael N. Hall und Prof. Markus Affolter. Basel, den 21.11.2006 Prof. Hans-Peter Hauri Dekan Summary The growth controlling mammalian Target of Rapamycin (mTOR) is a conserved Ser/Thr kinase found in two structurally and functionally distinct complexes, mTORC1 and mTORC2. The tumor suppressor TSC1-TSC2 complex inhibits mTORC1 by acting on the small GTPase Rheb, but the role of TSC1-TSC2 and Rheb in the regulation of mTORC2 is unclear. Here we examined the role of TSC1-TSC2 in the regulation of mTORC2 in human embryonic kidney 293 cells. Induced knockdown of TSC1 and TSC2 (TSC1/2) stimulated mTORC2-dependent actin cytoskeleton organization and Paxillin phosphorylation. Furthermore, TSC1/2 siRNA increased mTORC2-dependent Ser473 phosphorylation of plasma membrane bound, myristoylated Akt/PKB. This suggests that loss of Akt/PKB Ser473 phosphorylation in TSC mutant cells, as reported previously, is due to inhibition of Akt/PKB localization rather than inhibition of mTORC2 activity. Amino acids and overexpression of Rheb failed to stimulate mTORC2 signaling. Thus, TSC1-TSC2 also inhibits mTORC2, but possibly independently of Rheb. Our results suggest that mTORC2 hyperactivation may contribute to the pathophysiology of diseases such as cancer and Tuberous Sclerosis Complex. i Acknowledgement During my PhD studies in the Biozentrum I received a lot of support from many people around me who I mention here to express my gratefulness. -

The Atypical Guanine-Nucleotide Exchange Factor, Dock7, Negatively Regulates Schwann Cell Differentiation and Myelination
The Journal of Neuroscience, August 31, 2011 • 31(35):12579–12592 • 12579 Cellular/Molecular The Atypical Guanine-Nucleotide Exchange Factor, Dock7, Negatively Regulates Schwann Cell Differentiation and Myelination Junji Yamauchi,1,3,5 Yuki Miyamoto,1 Hajime Hamasaki,1,3 Atsushi Sanbe,1 Shinji Kusakawa,1 Akane Nakamura,2 Hideki Tsumura,2 Masahiro Maeda,4 Noriko Nemoto,6 Katsumasa Kawahara,5 Tomohiro Torii,1 and Akito Tanoue1 1Department of Pharmacology and 2Laboratory Animal Resource Facility, National Research Institute for Child Health and Development, Setagaya, Tokyo 157-8535, Japan, 3Department of Biological Sciences, Tokyo Institute of Technology, Midori, Yokohama 226-8501, Japan, 4IBL, Ltd., Fujioka, Gumma 375-0005, Japan, and 5Department of Physiology and 6Bioimaging Research Center, Kitasato University School of Medicine, Sagamihara, Kanagawa 252-0374, Japan In development of the peripheral nervous system, Schwann cells proliferate, migrate, and ultimately differentiate to form myelin sheath. In all of the myelination stages, Schwann cells continuously undergo morphological changes; however, little is known about their underlying molecular mechanisms. We previously cloned the dock7 gene encoding the atypical Rho family guanine-nucleotide exchange factor (GEF) and reported the positive role of Dock7, the target Rho GTPases Rac/Cdc42, and the downstream c-Jun N-terminal kinase in Schwann cell migration (Yamauchi et al., 2008). We investigated the role of Dock7 in Schwann cell differentiation and myelination. Knockdown of Dock7 by the specific small interfering (si)RNA in primary Schwann cells promotes dibutyryl cAMP-induced morpholog- ical differentiation, indicating the negative role of Dock7 in Schwann cell differentiation. It also results in a shorter duration of activation of Rac/Cdc42 and JNK, which is the negative regulator of myelination, and the earlier activation of Rho and Rho-kinase, which is the positive regulator of myelination. -

Mutation Analysis of Son of Sevenless in Juvenile Myelomonocytic Leukemia
Letters to the Editor 1108 3 Licht JD. Reconstructing a disease: what essential features of the the development of acute promyelocytic leukemia in transgenic retinoic acid receptor fusion oncoproteins generate acute promye- mice. Proc Natl Acad Sci USA 2000; 97: 13306–13311. locytic leukemia? Cancer Cell 2006; 9: 73–74. 10 Sternsdorf T, Phan VT, Maunakea ML, Ocampo CB, Sohal J, 4 Cools J, DeAngelo DJ, Gotlib J, Stover EH, Legare RD, Cortes J et al. Silletto A et al. Forced retinoic acid receptor alpha homodimers A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 prime mice for APL-like leukemia. Cancer Cell 2006; 9: 81–94. genes as a therapeutic target of imatinib in idiopathic hypereosino- 11 Kwok C, Zeisig BB, Dong S, So CW. Forced homo-oligomerization philic syndrome. N Engl J Med 2003; 348: 1201–1214. of RARalpha leads to transformation of primary hematopoietic 5 Kaufmann I, Martin G, Friedlein A, Langen H, Keller W. Human cells. Cancer Cell 2006; 9: 95–108. Fip1 is a subunit of CPSF that binds to U-rich RNA elements and 12 Palaniswamy V, Moraes KC, Wilusz CJ, Wilusz J. Nucleophosmin stimulates poly(A) polymerase. EMBO J 2004; 23: 616–626. is selectively deposited on mRNA during polyadenylation. Nat 6 Sainty D, Liso V, Cantu-Rajnoldi A, Head D, Mozziconacci MJ, Struct Mol Biol 2006; 13: 429–435. Arnoulet C et al. A new morphologic classification system for acute 13 Rego EM, Ruggero D, Tribioli C, Cattoretti G, Kogan S, Redner RL promyelocytic leukemia distinguishes cases with underlying PLZF/ et al. -

Figure S1. HAEC ROS Production and ML090 NOX5-Inhibition
Figure S1. HAEC ROS production and ML090 NOX5-inhibition. (a) Extracellular H2O2 production in HAEC treated with ML090 at different concentrations and 24 h after being infected with GFP and NOX5-β adenoviruses (MOI 100). **p< 0.01, and ****p< 0.0001 vs control NOX5-β-infected cells (ML090, 0 nM). Results expressed as mean ± SEM. Fold increase vs GFP-infected cells with 0 nM of ML090. n= 6. (b) NOX5-β overexpression and DHE oxidation in HAEC. Representative images from three experiments are shown. Intracellular superoxide anion production of HAEC 24 h after infection with GFP and NOX5-β adenoviruses at different MOIs treated or not with ML090 (10 nM). MOI: Multiplicity of infection. Figure S2. Ontology analysis of HAEC infected with NOX5-β. Ontology analysis shows that the response to unfolded protein is the most relevant. Figure S3. UPR mRNA expression in heart of infarcted transgenic mice. n= 12-13. Results expressed as mean ± SEM. Table S1: Altered gene expression due to NOX5-β expression at 12 h (bold, highlighted in yellow). N12hvsG12h N18hvsG18h N24hvsG24h GeneName GeneDescription TranscriptID logFC p-value logFC p-value logFC p-value family with sequence similarity NM_052966 1.45 1.20E-17 2.44 3.27E-19 2.96 6.24E-21 FAM129A 129. member A DnaJ (Hsp40) homolog. NM_001130182 2.19 9.83E-20 2.94 2.90E-19 3.01 1.68E-19 DNAJA4 subfamily A. member 4 phorbol-12-myristate-13-acetate- NM_021127 0.93 1.84E-12 2.41 1.32E-17 2.69 1.43E-18 PMAIP1 induced protein 1 E2F7 E2F transcription factor 7 NM_203394 0.71 8.35E-11 2.20 2.21E-17 2.48 1.84E-18 DnaJ (Hsp40) homolog. -

Identification of Two Signaling Submodules Within the Crkii/ELMO
Cell Death and Differentiation (2007) 14, 963–972 & 2007 Nature Publishing Group All rights reserved 1350-9047/07 $30.00 www.nature.com/cdd Identification of two signaling submodules within the CrkII/ELMO/Dock180 pathway regulating engulfment of apoptotic cells A-C Tosello-Trampont1,4, JM Kinchen1,2,4, E Brugnera1,3, LB Haney1, MO Hengartner2 and KS Ravichandran*,1 Removal of apoptotic cells is a dynamic process coordinated by ligands on apoptotic cells, and receptors and other signaling proteins on the phagocyte. One of the fundamental challenges is to understand how different phagocyte proteins form specific and functional complexes to orchestrate the recognition/removal of apoptotic cells. One evolutionarily conserved pathway involves the proteins cell death abnormal (CED)-2/chicken tumor virus no. 10 (CT10) regulator of kinase (Crk)II, CED-5/180 kDa protein downstream of chicken tumor virus no. 10 (Crk) (Dock180), CED-12/engulfment and migration (ELMO) and MIG-2/RhoG, leading to activation of the small GTPase CED-10/Rac and cytoskeletal remodeling to promote corpse uptake. Although the role of ELMO : Dock180 in regulating Rac activation has been well defined, the function of CED-2/CrkII in this complex is less well understood. Here, using functional studies in cell lines, we observe that a direct interaction between CrkII and Dock180 is not required for efficient removal of apoptotic cells. Similarly, mutants of CED-5 lacking the CED-2 interaction motifs could rescue engulfment and migration defects in CED-5 deficient worms. Mutants of CrkII and Dock180 that could not biochemically interact could colocalize in membrane ruffles. -

Disassembly of the Dying: Mechanisms and Functions
Review Disassembly of the Dying: Mechanisms and Functions 1 1, Georgia K. Atkin-Smith and Ivan K.H. Poon * The disassembly of an apoptotic cell into subcellular fragments, termed Trends apoptotic bodies (ApoBDs), is a hallmark of apoptosis. Although the genera- Apoptotic cell disassembly is a highly tion of ApoBDs is generally understood as being stochastic, it is becoming complex process regulated by a series of well-coordinated morphological increasingly clear that ApoBD formation is a highly regulated process involv- steps including apoptotic membrane ing distinct morphological steps and molecular factors. Functionally, ApoBDs blebbing, apoptotic protrusion forma- fi could facilitate the ef cient clearance of apoptotic material by surrounding tion, and fragmentation. phagocytes as well as mediate the transfer of biomolecules including micro- Plasma-membrane blebbing is not the RNAs and proteins between cells to aid in intercellular communications. sole process required for apoptotic Therefore, the formation of ApoBDs is an important process downstream body (ApoBD) formation, but mem- brane protrusions including microtu- from apoptotic cell death. We discuss here the mechanisms and functions bule spikes, apoptopodia, and of apoptotic cell disassembly. beaded apoptopodia may act in con- cert to aid the generation of ApoBDs. Cell Disassembly as a Key Downstream Process of Apoptotic Cell Death The mechanism of how apoptotic cells Billions of cells undergo apoptosis (a form of programmed cell death) daily as part of disassembly can determine the quantity and quality (size and contents) of physiological homeostasis [1]. At later stages of apoptosis, some cell types can generate ApoBDs. subcellular (1–5 mm) membrane-bound extracellular vesicles termed apoptotic bodies – (ApoBDs, see Glossary) [1 3]. -

Snapshot: Axon Guidance II Alex L
SnapShot: Axon Guidance II Alex L. Kolodkin1 and R. Jeroen Pasterkamp2 1Department of Neuroscience, HHMI, The Johns Hopkins University School of Medicine, Baltimore, MD 21212, USA 2Department of Neuroscience and Pharmacology, Rudolf Magnus Institute of Neuroscience, University Medical Center Utrecht, 3584 CG Utrecht, The Netherlands Axon guidance protein Chemotrophic effect Ligand-binding receptor Receptor processing Coreceptor Receptor signaling COFILIN, LIMK1, PI3K, BMP SE Repulsion BMP-RIB, BMP-RII - - SMAD1, SMAD6 DRAXIN SE Repulsion DCC - - - a2-CHIMAERIN, EPHEXIN-1, EPHRINA GPI Repulsion EPHA ADAM10 - NCK1/2, RAC1, RHOA, SPAR, VAV2/3 NCK1, PAK, p120GAP, RHOA, EPHRINB TM Repulsion EPHB MMP2/9 - ROCK, VAV2/3 TM Repulsion EPHRINA ADAM10 P75 FYN EPHA Attraction EPHRINA - RET - CDC42, DOCK180, GRB4, NCK2, EPHB TM Repulsion EPHRINB ADAM10, PS1 - PAK, RAC1 FGF SE Attraction FGFR1 - - - CDC42, DOCK180, ENA/VASP, APP, HSPG, ERK1/2, FAK, FYN, NCK1, N- SE Attraction DCC ADAM10, PS1 ROBO1 WASP, PAK, PI3K, PIP2, PKC, NETRIN RAC1, RHOA, TRIO, TRP Attraction NEOGENIN - - - Repulsion UNC5 - (DCC) FAK, SHP2, SRC FAK, LARG, LMO4, MYOIIA, RGM GPI Repulsion NEOGENIN TACE, PS1 UNC5 p120GAP, PKC, RAS, RHOA PLEXINA TM Repulsion SEMA1a - - PEBBLE, RHO, p190RHOGAP 14-3-3e, GYC76C, MICAL, SEMA1 TM Repulsion PLEXINA - OTK NERVY, PKA SE Repulsion PLEXINB - - RAC, RHO SEMA2 Attraction PLEXINB - - - AKT, COFILIN, CDK5, CRMP, FARP, NRP1/2, CAMs, SE Repulsion PLEXINA1-4 CALPAIN1 FYN, GSK3b, LIMK1, PI3K, RAC1, RTKs, ROBO SEMA3 RAP1, RAS, RND Attraction - - NRP1/2, -

Rho Guanine Nucleotide Exchange Factors: Regulators of Rho Gtpase Activity in Development and Disease
Oncogene (2014) 33, 4021–4035 & 2014 Macmillan Publishers Limited All rights reserved 0950-9232/14 www.nature.com/onc REVIEW Rho guanine nucleotide exchange factors: regulators of Rho GTPase activity in development and disease DR Cook1, KL Rossman2,3 and CJ Der1,2,3 The aberrant activity of Ras homologous (Rho) family small GTPases (20 human members) has been implicated in cancer and other human diseases. However, in contrast to the direct mutational activation of Ras found in cancer and developmental disorders, Rho GTPases are activated most commonly in disease by indirect mechanisms. One prevalent mechanism involves aberrant Rho activation via the deregulated expression and/or activity of Rho family guanine nucleotide exchange factors (RhoGEFs). RhoGEFs promote formation of the active GTP-bound state of Rho GTPases. The largest family of RhoGEFs is comprised of the Dbl family RhoGEFs with 70 human members. The multitude of RhoGEFs that activate a single Rho GTPase reflects the very specific role of each RhoGEF in controlling distinct signaling mechanisms involved in Rho activation. In this review, we summarize the role of Dbl RhoGEFs in development and disease, with a focus on Ect2 (epithelial cell transforming squence 2), Tiam1 (T-cell lymphoma invasion and metastasis 1), Vav and P-Rex1/2 (PtdIns(3,4,5)P3 (phosphatidylinositol (3,4,5)-triphosphate)-dependent Rac exchanger). Oncogene (2014) 33, 4021–4035; doi:10.1038/onc.2013.362; published online 16 September 2013 Keywords: Rac1; RhoA; Cdc42; guanine nucleotide exchange factors; cancer; -

Structural Analysis of Autoinhibition in the Ras-Specific Exchange Factor
RESEARCH ARTICLE elife.elifesciences.org Structural analysis of autoinhibition in the Ras-specific exchange factor RasGRP1 Jeffrey S Iwig1,2, Yvonne Vercoulen3†, Rahul Das1,2†, Tiago Barros1,2,4, Andre Limnander3, Yan Che1,2, Jeffrey G Pelton2, David E Wemmer2,5,6, Jeroen P Roose3*, John Kuriyan1,2,4,5,6* 1Department of Molecular and Cell Biology, University of California, Berkeley, Berkeley, United States; 2California Institute for Quantitative Biosciences, University of California, Berkeley, Berkeley, United States; 3Department of Anatomy, University of California, San Francisco, San Francisco, United States; 4Howard Hughes Medical Institute, University of California, Berkeley, Berkeley, United States; 5Department of Chemistry, University of California, Berkeley, Berkeley, United States; 6Physical Biosciences Division, Lawrence Berkeley National Laboratory, Berkeley, United States Abstract RasGRP1 and SOS are Ras-specific nucleotide exchange factors that have distinct roles in lymphocyte development. RasGRP1 is important in some cancers and autoimmune diseases but, in contrast to SOS, its regulatory mechanisms are poorly understood. Activating signals lead to the membrane recruitment of RasGRP1 and Ras engagement, but it is unclear how interactions between RasGRP1 and Ras are suppressed in the absence of such signals. We present a crystal structure of a fragment of RasGRP1 in which the Ras-binding site is blocked by an interdomain linker and the membrane-interaction surface of RasGRP1 is hidden within a dimerization interface that may be stabilized by the C-terminal oligomerization domain. NMR data demonstrate that calcium binding to the regulatory module generates substantial conformational changes that are incompatible with the inactive assembly. These features allow RasGRP1 to be maintained in an inactive state that is poised for activation by calcium and membrane-localization signals. -

Egfr Activates a Taz-Driven Oncogenic Program in Glioblastoma
EGFR ACTIVATES A TAZ-DRIVEN ONCOGENIC PROGRAM IN GLIOBLASTOMA by Minling Gao A thesis submitted to Johns Hopkins University in conformity with the requirements for the degree of Doctor of Philosophy Baltimore, Maryland March 2020 ©2020 Minling Gao All rights reserved Abstract Hyperactivated EGFR signaling is associated with about 45% of Glioblastoma (GBM), the most aggressive and lethal primary brain tumor in humans. However, the oncogenic transcriptional events driven by EGFR are still incompletely understood. We studied the role of the transcription factor TAZ to better understand master transcriptional regulators in mediating the EGFR signaling pathway in GBM. The transcriptional coactivator with PDZ- binding motif (TAZ) and its paralog gene, the Yes-associated protein (YAP) are two transcriptional co-activators that play important roles in multiple cancer types and are regulated in a context-dependent manner by various upstream signaling pathways, e.g. the Hippo, WNT and GPCR signaling. In GBM cells, TAZ functions as an oncogene that drives mesenchymal transition and radioresistance. This thesis intends to broaden our understanding of EGFR signaling and TAZ regulation in GBM. In patient-derived GBM cell models, EGF induced TAZ and its known gene targets through EGFR and downstream tyrosine kinases (ERK1/2 and STAT3). In GBM cells with EGFRvIII, an EGF-independent and constitutively active mutation, TAZ showed EGF- independent hyperactivation when compared to EGFRvIII-negative cells. These results revealed a novel EGFR-TAZ signaling axis in GBM cells. The second contribution of this thesis is that we performed next-generation sequencing to establish the first genome-wide map of EGF-induced TAZ target genes.