Hypertrophic Cardiomyopathy Testing
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Pathogenic Variant Identified
PERFORMED AT PHOSPHORUS DIAGNOSTICS, 400 PLAZA DRIVE, SUITE 401, SECAUCUS, NJ 07094 ORDERING PRACTICE JANE DOE TEST INFORMATION Practice Code: 100 DOB: 1973-02-19 Panel: Pan Arrhythmia and Cardiomyopathy Panel Sample Cardiology Clinic Gender: Female Indication: Diagnostic for Patient History 374 Broadway Ethnicity: European New York, NY 10001 Procedure ID: 87000 Physician: Dr. Sample Kit Barcode: 201612092248585 Specimen: Blood, #10000 Specimen Collected: 2016-01-12 Specimen Received: 2016-01-13 Specimen Analyzed: 2016-01-21 Report Generated: 2016-02-03 SUMMARY OF RESULTS Positive: Pathogenic Variant Identified GENE LOCATION VARIANT INHERITANCE ZYGOSITY CLASSIFICATION KCNQ1 Exon 6; Chrom 11 c.914G>A Autosomal Dominant Heterozygous PATHOGENIC (p.Trp205Ter) MYH7 Exon 38; Chrom 14 c.26647G>A Unknown Heterozygous UNCERTAIN (p.Glu1883Lys) SIGNIFICANCE INTERPRETATION This patient is heterozygous for the c.914G>A (p.Trp205Ter) pathogenic variant in KCNQ1. This result indicates that this patient may be affected with, or predisposed to developing, long QT syndrome. This patient is also heterozygous for the c.185G>A (p.Lys65Ala) variant of unknown clinical significance in MYH7. The contribution of this variant to the patient’s phenotype is unknown. RECOMMENDATIONS Clinical follow-up with a cardiologist or cardiogeneticist is recommended to discuss medical management. Genetic counseling is recommended for this patient. Genetic testing may be useful in identifying family members at risk for disease. Genetic counseling is recommended for all individuals undergoing genetic testing. VARIANT INFORMATION PATHOGENIC KCNQ1 c.914G>A (p.Trp305Ter) This variant leads to a premature termination at codon 305, which is expected to result in an absent or significantly disrupted protein product. -

Danon Disease Dr
Danon disease Dr. Andrés R. Pérez Riera Danon disease Other names: glycogen storage disease type 2B; glycogen storage disease type IIb; lysosomal glycogen storage disease with normal acid maltase Danon disease is a condition characterized by cardiomyopathy; weakening of the muscles used for movement, called skeletal muscles, (myopathy); and intellectual disability. Males with Danon disease usually develop the condition earlier than females and are more severely affected. Signs and symptoms begin in childhood or adolescence in most affected males and in early adulthood in most affected females. Affected males, on average, live to age 19, while affected females live to an average age of 34. Cardiomyopathy is the most common symptom of Danon disease and occurs in all males with the condition. Most affected men have hypertrophic cardiomyopathy, which is a thickening of the heart muscle that may make it harder for the heart to pump blood. Other affected males have dilated cardiomyopathy, which is a condition that weakens and enlarges the heart, preventing it from pumping blood efficiently. Some affected men with hypertrophic cardiomyopathy later develop dilated cardiomyopathy. Either type of cardiomyopathy can lead to heart failure and premature death. Most women with Danon disease also develop cardiomyopathy; of the women who have this feature, about half have hypertrophic cardiomyopathy, and the other half have dilated cardiomyopathy. Affected individuals can have other heart-related signs and symptoms, including a sensation of fluttering or pounding in the chest (palpitations), arrhythmias, or chest pain. Many affected individuals have conduction abnormalities. People with Danon disease are often affected by a specific conduction abnormality known as cardiac preexcitation type Wolff-Parkinson White. -

6 the Glycogen Storage Diseases and Related Disorders
6 The Glycogen Storage Diseases and Related Disorders G. Peter A. Smit, Jan Peter Rake, Hasan O. Akman, Salvatore DiMauro 6.1 The Liver Glycogenoses – 103 6.1.1 Glycogen Storage Disease Type I (Glucose-6-Phosphatase or Translocase Deficiency) – 103 6.1.2 Glycogen Storage Disease Type III (Debranching Enzyme Deficiency) – 108 6.1.3 Glycogen Storage Disease Type IV (Branching Enzyme Deficiency) – 109 6.1.4 Glycogen Storage Disease Type VI (Glycogen Phosphorylase Deficiency) – 111 6.1.5 Glycogen Storage Disease Type IX (Phosphorylase Kinase Deficiency) – 111 6.1.6 Glycogen Storage Disease Type 0 (Glycogen Synthase Deficiency) – 112 6.2 The Muscle Glycogenoses – 112 6.2.1 Glycogen Storage Disease Type V (Myophosphorylase Deficiency) – 113 6.2.2 Glycogen Storage Disease Type VII (Phosphofructokinase Deficiency) – 113 6.2.3 Phosphoglycerate Kinase Deficiency – 114 6.2.4 Glycogen Storage Disease Type X (Phosphoglycerate Mutase Deficiency) – 114 6.2.5 Glycogen Storage Disease Type XII (Aldolase A Deficiency) – 114 6.2.6 Glycogen Storage Disease Type XIII (E-Enolase Deficiency) – 115 6.2.7 Glycogen Storage Disease Type XI (Lactate Dehydrogenase Deficiency) – 115 6.2.8 Muscle Glycogen Storage Disease Type 0 (Glycogen Synthase Deficiency) – 115 6.3 The Generalized Glycogenoses and Related Disorders – 115 6.3.1 Glycogen Storage Disease Type II (Acid Maltase Deficiency) – 115 6.3.2 Danon Disease – 116 6.3.3 Lafora Disease – 116 References – 116 102 Chapter 6 · The Glycogen Storage Diseases and Related Disorders Glycogen Metabolism Glycogen is a macromolecule composed of glucose viding glucose and glycolytic intermediates (. Fig. 6.1). units. -

Danon Disease
Xu et al. Diagnostic Pathology (2021) 16:39 https://doi.org/10.1186/s13000-021-01100-8 CASE REPORT Open Access Danon disease: a case report and literature review Jiamin Xu1†, Zhu Li1†, Yihai Liu1†, Xinlin Zhang1, Fengnan Niu2, Hongyan Zheng1, Lian Wang1, Lina Kang1* , Kun Wang1* and Biao Xu1* Abstract Background: Danon disease (DD) is a rare x-linked dominant multisystemic disorder with a clinical triad of severe cardiomyopathy, skeletal myopathy, and mental retardation. It is caused by a defect in the lysosomal-associated membrane protein-2 (LAMP2) gene, which leads to the formation of autophagic vacuoles containing glycogen granule deposits in skeletal and cardiac muscle fibers. So far, more than 50 different mutations in LAMP2 have been identified. Case presentation: Here, we report an 18-year-old male patient who was hospitalized for heart failure. Biopsy of the left lateral femoral muscle revealed scattered autophagic vacuoles in the muscle fibers with increased glycogen. Next generation sequencing (NGS) was used to detect gene mutations of the proband sample and a novel frameshift mutation (c.1052delG) has been identified in exon 8 of LAMP2, which leads to truncation of the protein. Conclusion: We found a novel frameshift mutation, a hemizygous mutation (c.1052delG) in exon 8 of LAMP2, identified as presenting the hypertrophic cardiomyopathy (HCM) phenotype. Genetic analysis is the gold standard for the diagnosis of DD and is essential to determine appropriate treatment strategies and to confirm the genetic risk of family members. Keywords: Danon disease, Cardiomyopathy, LAMP2, Mutation, NGS Background maturation. Autophagy is a very important biological DD is a rare x-linked dominant multisystemic disorder phenomenon involved in regulating the metabolic balance with clinical manifestations of severe cardiomyopathy, between the synthesis, degradation and reuse of cellular skeletal myopathy, and mental retardation. -

Genetic Testing Medical Policy – Genetics
Genetic Testing Medical Policy – Genetics Please complete all appropriate questions fully. Suggested medical record documentation: • Current History & Physical • Progress Notes • Family Genetic History • Genetic Counseling Evaluation *Failure to include suggested medical record documentation may result in delay or possible denial of request. PATIENT INFORMATION Name: Member ID: Group ID: PROCEDURE INFORMATION Genetic Counseling performed: c Yes c No **Please check the requested analyte(s), identify number of units requested, and provide indication/rationale for testing. 81400 Molecular Pathology Level 1 Units _____ c ACADM (acyl-CoA dehydrogenase, C-4 to C-12 straight chain, MCAD) (e.g., medium chain acyl dehydrogenase deficiency), K304E variant _____ c ACE (angiotensin converting enzyme) (e.g., hereditary blood pressure regulation), insertion/deletion variant _____ c AGTR1 (angiotensin II receptor, type 1) (e.g., essential hypertension), 1166A>C variant _____ c BCKDHA (branched chain keto acid dehydrogenase E1, alpha polypeptide) (e.g., maple syrup urine disease, type 1A), Y438N variant _____ c CCR5 (chemokine C-C motif receptor 5) (e.g., HIV resistance), 32-bp deletion mutation/794 825del32 deletion _____ c CLRN1 (clarin 1) (e.g., Usher syndrome, type 3), N48K variant _____ c DPYD (dihydropyrimidine dehydrogenase) (e.g., 5-fluorouracil/5-FU and capecitabine drug metabolism), IVS14+1G>A variant _____ c F13B (coagulation factor XIII, B polypeptide) (e.g., hereditary hypercoagulability), V34L variant _____ c F2 (coagulation factor 2) (e.g., -
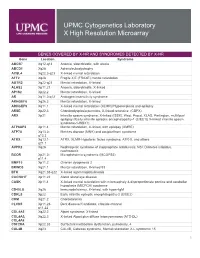
Genes Covered and Disorders Detected by X-HR Microarray
UPMC Cytogenetics Laboratory X High Resolution Microarray GENES COVERED BY X-HR AND SYNDROMES DETECTED BY X-HR Gene Location Syndrome ABCB7 Xq12-q13 Anemia, sideroblastic, with ataxia ABCD1 Xq28 Adrenoleukodystrophy ACSL4 Xq22.3-q23 X-linked mental retardation AFF2 Xq28 Fragile X E (FRAXE) mental retardation AGTR2 Xq22-q23 Mental retardation, X-linked ALAS2 Xp11.21 Anemia, sideroblastic, X-linked AP1S2 Xp22.2 Mental retardation, X-linked AR Xq11.2-q12 Androgen insensitivity syndrome ARHGEF6 Xq26.3 Mental retardation, X-linked ARHGEF9 Xq11.1 X-linked mental retardation (XLMR)//Hyperekplexia and epilepsy ARSE Xp22.3 Chondrodysplasia punctata, X-linked recessive (CDPX) ARX Xp21 Infantile spasm syndrome, X-linked (ISSX), West, Proud, XLAG, Partington, multifocal epilepsy //Early infantile epileptic encephalopathy-1 (EIEE1)( X-linked infantile spasm syndrome-1-ISSX1) ATP6AP2 Xp11.4 Mental retardation, X-linked, with epilepsy (XMRE) ATP7A Xq13.2- Menkes disease (MNK) and occipital horn syndrome q13.3 ATRX Xq13.1- ATRX, XLMR-Hypotonic facies syndrome, ATR-X, and others q21.1 AVPR2 Xq28 Nephrogenic syndrome of inappropriate antidiuresis; NSI; Diabetes insipidus, nephrogenic BCOR Xp21.2- Microphthalmia syndromic (MCOPS2) p11.4 BMP15 Xp11.2 Ovarian dysgenesis 2 BRWD3 Xq21.1 Mental retardation, X-linked 93 BTK Xq21.33-q22 X-linked agammaglobulinemia CACNA1F Xp11.23 Aland Island eye disease CASK Xp11.4 X-linked mental retardation with microcephaly & disproportionate pontine and cerebellar hypoplasia (MICPCH) syndrome CD40LG Xq26 Immunodeficiency, -

Generation of Induced Pluripotent Stem Cells from a Female Patient with a Xq27.3-Q28 Deletion to Establish Disease Models and Identify Therapies
CELLULAR REPROGRAMMING Volume 22, Number 4, 2020 Research Article ª Mary Ann Liebert, Inc. DOI: 10.1089/cell.2020.0012 Generation of Induced Pluripotent Stem Cells from a Female Patient with a Xq27.3-q28 Deletion to Establish Disease Models and Identify Therapies Noriko Watanabe,1,* Kohei Kitada,1,* Katherine E. Santostefano,1 Airi Yokoyama,1 Sara M. Waldrop,1 Coy D. Heldermon,2 Daisuke Tachibana,3 Masayasu Koyama,3 Amy M. Meacham,2 Christina A. Pacak,4 and Naohiro Terada1 Abstract Since it is extremely difficult to establish an animal model for human chromosomal abnormalities, induced pluripotent stem cells (iPSCs) provide a powerful alternative to study underlying mechanisms of these disorders and identify potential therapeutic interventions. In this study we established iPSCs from a young girl with a hemizygous deletion of Xq27.3-q28 who exhibited global developmental delay and intellectual disability from early in infancy. The deletion site on the X chromosome includes Fragile X Mental Retardation 1 (FMR1), the gene responsible for fragile X syndrome, which likely contributes to the patient’s neurodevelopmental ab- normalities. The FMR1 gene was expressed in approximately half of the iPSC clones we generated while it was absent in the other half due to the random inactivation of normal and abnormal X chromosomes. The normal or absent expression pattern of the FMR1 gene was not altered when the iPSCs were differentiated into neural progenitor cells (NPCs). Moreover, chromosome reactivating reagents such as 5-aza-2-deoxycytidine, tri- chostatin A, and UNC0638, were tested in an attempt to reactivate the suppressed FMR1 gene in affected iPSC- NPCs. -

Clinical Manifestation in Females with X-Linked Metabolic Disorders
Review Journal of Inborn Errors Clinical Manifestation in Females with of Metabolism & Screening 2021, Volume 9: e20200024 X-linked Metabolic Disorders: Genetic and DOI: https://doi.org/10.1590/2326-4594- JIEMS-2020-0024 Pathophysiological Considerations 1 Michael Beck Abstract Inborn errors of metabolism are predominantly autosomal-recessive disorders, but several follow an X-linked pattern of inheritance. They are called X-linked recessive, if the female carriers are asymptomatic, and are called X-linked dominant disorders, if almost all females are affected. Conditions, in which some females have symptoms while others are asymptomatic lifelong are simply referred to as X-linked. The aim of this review is to point out the variability in clinical manifestation of affected females in some X-linked metabolic disorders and to discuss on the basis of these examples possible mechanisms that may explain the broad phenotypic spectrum, such as the type of the underlying mutation, the issue of autonomous versus non-autonomous gene expression and the degree of skewing of X-inactivation. The use of the terms “X-linked dominant” and “X-linked recessive” will be discussed. Keywords X-linked disorders, metabolic disorders, X-inactivation, pyruvate dehydrogenase, creatine transporter, cholesterol biosynthesis, hypophosphatemic rickets, lysosomal storage disorder. Introduction Since blood cells have a very great number of cell divisions, even a slight selection against the normal allele in blood cells In X-linked disorders an important, but not only, factor for may lead to a non-random X inactivation pattern in these cells. disease expression in heterozygous patients is the degree of X-inactivation, however, is not complete, in female cells X-inactivation skewing. -

Danon's Disease As a Cause of Hypertrophic
842 CARDIOVASCULAR MEDICINE Heart: first published as 10.1136/hrt.2003.029090 on 14 July 2004. Downloaded from Danon’s disease as a cause of hypertrophic cardiomyopathy: a systematic survey P Charron, E Villard, P Se´billon, P Laforeˆt, T Maisonobe, L Duboscq-Bidot, N Romero, V Drouin- Garraud, T Fre´bourg, P Richard, B Eymard, M Komajda ............................................................................................................................... Heart 2004;90:842–846. doi: 10.1136/hrt.2003.029504 Background: Hypertrophic cardiomyopathy (HCM) is an autosomal dominant disease caused by mutations in sarcomeric genes. However, extensive genetic screening failed to identify a mutation in about a third of cases. One possible explanation is that other diseases, caused by other genes, may mimic HCM. Objective: To investigate the possible involvement of Danon’s disease, an X linked lysosomal disease, in a See end of article for large population of patients with HCM. authors’ affiliations Methods: A population of 197 index cases was considered; 124 were subsequently excluded because of a ....................... mutation in sarcomeric genes and 23 because of autosomal dominant inheritance. Fifty index cases were Correspondence to: therefore included in molecular analysis (direct sequencing) of the lysosome associated membrane protein Dr Philippe Charron, 2 (LAMP2) gene responsible for Danon’s disease. De´partement de Results: Two new mutations leading to premature stop codons were identified in patients who evolved Ge´ne´tique, Batiment towards severe heart failure (, 25 years old): 657C.T and 173_179del. The prevalence was therefore Babinski, Hoˆpital Pitie´- Salpeˆtrie`re, 47 Blvd de 1% of the total population (two of 197) or 4% of enrolled index cases (two of 50). -

Danon Disease: Gender Differences in Presentation and Outcomes
International Journal of Cardiology 286 (2019) 92–98 Contents lists available at ScienceDirect International Journal of Cardiology journal homepage: www.elsevier.com/locate/ijcard Danon disease: Gender differences in presentation and outcomes Michela Brambatti a,1,OrenCaspia,1,AlessandroMaoloa, Elliott Koshi a, Barry Greenberg a, Matthew R.G. Taylor b,EricD.Adlera,⁎,2 a Division of Cardiology, University of California San Diego, San Diego, CA, USA b Cardiovascular Institute and Adult Medical Genetics Program, University of Colorado Denver, CO, USA article info abstract Article history: Background: Danon disease (DD) is a rare X-linked autophagic vacuolar myopathy, characterized by high Received 7 December 2018 penetrance and severe cardiomyopathy. Because of its rarity, the natural history (NH) is uncertain. Accepted 2 January 2019 Objectives: We aimed to describe disease variability and outcomes through a systematic review of all published Available online 16 February 2019 DD cases. Methods: Among 83 manuscripts in MEDLINE and EMBASE on DD cases published until October 2017, we Keywords: identified 146 patients with positive genetic testing for DD or positive muscle biopsy in a relative of a genetically Danon disease diagnosed proband. LAMP2 mutation fi Hypertrophic cardiomyopathy Results: 56 females and 90 males were identi ed. 92.5% of patients had cardiac abnormalities. Females presented Dilated cardiomyopathy with either hypertrophic cardiomyopathy (HCM, 70.3%) or dilated cardiomyopathy (DCM, 29.3%) whereas males Genetic testing presented with HCM 96.2% of the time. The composite outcome of death, heart transplant or ventricular assist Gender devices occurred equally in both sexes (32% of females and 37% of males, p = 0.60) but later in females (median age 38 years) than in males (median age 21 years, p b 0.001). -

Lysosomal Storage Disease Panel by Next-Generation Sequencing
TEST ID: LSDP LYSOSOMAL STORAGE DISEASE PANEL BY NEXT-GENERATION SEQUENCING USEFUL FOR REFERENCE VALUES } Follow up for abnormal biochemical results and confirmation of suspected lysosomal storage An interpretive report will be disease (LSD) provided. } Identifying mutations within genes known to be associated with lysosomal storage disease, allowing for predictive testing of at-risk family members ANALYTIC TIME 4 weeks CLINICAL INFORMATION Lysosomal storage diseases (LSDs) encompass a group of over 40 inherited biochemical diseases in which genetic mutations cause defective lysosomal functioning. Lysosomes perform catabolic functions for cells, which is accomplished through activity of various proteins such as lysosomal enzymes, transport proteins, and other proteins. Functional deficits in these proteins cause an accumulation of substrates in cells leading to progressive organ dysfunction. This leads to variable clinical features that can affect the cardiovascular, neurological, ocular, and skeletal systems, among others. Clinical features are dependent on the amount and location of the substrate accumulation, but may include the following: characteristic facial features (coarse features), hepatomegaly, deafness, vision loss, abnormal skeletal findings, hydrops fetalis, ataxia, hypotonia, developmental delay/regression, and intellectual disability. Age of onset is variable, with symptoms presenting from the prenatal period to adulthood, but generally LSDs are progressive and cause significant morbidity and mortality with a decreased lifespan. Enzyme replacement therapy and oral substrate inhibitors are therapeutic options for some LSDs. LSDs are inherited in an autosomal recessive manner with the exception of Hunter, Fabry, and Danon diseases, which are X-linked. There are some founder mutations associated with particular LSDs in the Ashkenazi Jewish and Finnish populations, leading to an increased carrier frequency for some. -

Neurodevelopment Next-Generation
NEURODEVELOPMENT NEXT-GENERATION SEQUENCING PANELS Mail: One Gustave L. Levy Place, Box 1497 CLIA #: 33D2097541 Specimens: 1428 Madison Ave, Atran Bldg, T: 800-298-6470 Page 1 of 125 Rm 2-25 New York, NY 10029 F: 212-241-0139 www.sema4genomics.com Mail: One Gustave L. Levy Place, Box 1497 CLIA #: 33D2097541 Specimens: 1428 Madison Ave, Atran Bldg, T: 800-298-6470 Page 2 of 125 Rm 2-25 New York, NY 10029 F: 212-241-0139 www.sema4genomics.com Mail: One Gustave L. Levy Place, Box 1497 CLIA #: 33D2097541 Specimens: 1428 Madison Ave, Atran Bldg, T: 800-298-6470 Page 3 of 125 Rm 2-25 New York, NY 10029 F: 212-241-0139 www.sema4genomics.com TABLE OF CONTENTS GENETIC TESTING FOR NEURODEVELOPMENTAL DISEASE 5 GENETICS 5 INDICATIONS 5 TESTING METHODS, SENSITIVITY, AND LIMITATIONS 6 TURNAROUND TIME 8 SPECIMEN AND SHIPPING REQUIREMENTS 8 CUSTOMER SERVICES AND GENETIC COUNSELING 9 THE COMPREHENSIVE EPILEPSY AND AUTISM PANEL 10 THE COMPREHENSIVE EPILEPSY PANEL 32 FOCAL, GENERALIZED, AND MYOCLONIC EPILEPSY SUBPANEL 48 INFANTILE EPILEPSY SUBPANEL 52 MIGRAINE SUBPANEL 56 NEURONAL CEROID LIPOFUSCINOSES SUBPANEL 57 NEURONAL MIGRATION SUBPANEL 58 SYNDROMIC EPILEPSY AND INTELLECTUAL DISABILITY SUBPANEL 61 THE COMPREHENSIVE AUTISM SPECTRUM DISORDER PANEL 67 STAT AUTISM SPECTRUM DISORDER SUBPANEL 80 MICROCEPHALY PANEL 84 REFERENCES 89 DISCLAIMER 108 Mail: One Gustave L. Levy Place, Box 1497 CLIA #: 33D2097541 Specimens: 1428 Madison Ave, Atran Bldg, T: 800-298-6470 Page 4 of 125 Rm 2-25 New York, NY 10029 F: 212-241-0139 www.sema4genomics.com Genetic Testing for NEURODEVELOPMENTAL DISEASE Sema4 offers targeted next-generation sequencing (NGS) for neurodevelopmental conditions such as autism spectrum disorder, Intellectual disability, epilepsy, seizures, brain malformations, and microcephaly.