Pathogenic Variant Identified
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Case of Prenatal Diagnosis of 18P Deletion Syndrome Following
Zhao et al. Molecular Cytogenetics (2019) 12:53 https://doi.org/10.1186/s13039-019-0464-y CASE REPORT Open Access A case of prenatal diagnosis of 18p deletion syndrome following noninvasive prenatal testing Ganye Zhao, Peng Dai, Shanshan Gao, Xuechao Zhao, Conghui Wang, Lina Liu and Xiangdong Kong* Abstract Background: Chromosome 18p deletion syndrome is a disease caused by the complete or partial deletion of the short arm of chromosome 18, there were few cases reported about the prenatal diagnosis of 18p deletion syndrome. Noninvasive prenatal testing (NIPT) is widely used in the screening of common fetal chromosome aneuploidy. However, the segmental deletions and duplications should also be concerned. Except that some cases had increased nuchal translucency or holoprosencephaly, most of the fetal phenotype of 18p deletion syndrome may not be evident during the pregnancy, 18p deletion syndrome was always accidentally discovered during the prenatal examination. Case presentations: In our case, we found a pure partial monosomy 18p deletion during the confirmation of the result of NIPT by copy number variation sequencing (CNV-Seq). The result of NIPT suggested that there was a partial or complete deletion of X chromosome. The amniotic fluid karyotype was normal, but result of CNV-Seq indicated a 7.56 Mb deletion on the short arm of chromosome 18 but not in the couple, which means the deletion was de novo deletion. Finally, the parents chose to terminate the pregnancy. Conclusions: To our knowledge, this is the first case of prenatal diagnosis of 18p deletion syndrome following NIPT.NIPT combined with ultrasound may be a relatively efficient method to screen chromosome microdeletions especially for the 18p deletion syndrome. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in -

Danon Disease Dr
Danon disease Dr. Andrés R. Pérez Riera Danon disease Other names: glycogen storage disease type 2B; glycogen storage disease type IIb; lysosomal glycogen storage disease with normal acid maltase Danon disease is a condition characterized by cardiomyopathy; weakening of the muscles used for movement, called skeletal muscles, (myopathy); and intellectual disability. Males with Danon disease usually develop the condition earlier than females and are more severely affected. Signs and symptoms begin in childhood or adolescence in most affected males and in early adulthood in most affected females. Affected males, on average, live to age 19, while affected females live to an average age of 34. Cardiomyopathy is the most common symptom of Danon disease and occurs in all males with the condition. Most affected men have hypertrophic cardiomyopathy, which is a thickening of the heart muscle that may make it harder for the heart to pump blood. Other affected males have dilated cardiomyopathy, which is a condition that weakens and enlarges the heart, preventing it from pumping blood efficiently. Some affected men with hypertrophic cardiomyopathy later develop dilated cardiomyopathy. Either type of cardiomyopathy can lead to heart failure and premature death. Most women with Danon disease also develop cardiomyopathy; of the women who have this feature, about half have hypertrophic cardiomyopathy, and the other half have dilated cardiomyopathy. Affected individuals can have other heart-related signs and symptoms, including a sensation of fluttering or pounding in the chest (palpitations), arrhythmias, or chest pain. Many affected individuals have conduction abnormalities. People with Danon disease are often affected by a specific conduction abnormality known as cardiac preexcitation type Wolff-Parkinson White. -

6 the Glycogen Storage Diseases and Related Disorders
6 The Glycogen Storage Diseases and Related Disorders G. Peter A. Smit, Jan Peter Rake, Hasan O. Akman, Salvatore DiMauro 6.1 The Liver Glycogenoses – 103 6.1.1 Glycogen Storage Disease Type I (Glucose-6-Phosphatase or Translocase Deficiency) – 103 6.1.2 Glycogen Storage Disease Type III (Debranching Enzyme Deficiency) – 108 6.1.3 Glycogen Storage Disease Type IV (Branching Enzyme Deficiency) – 109 6.1.4 Glycogen Storage Disease Type VI (Glycogen Phosphorylase Deficiency) – 111 6.1.5 Glycogen Storage Disease Type IX (Phosphorylase Kinase Deficiency) – 111 6.1.6 Glycogen Storage Disease Type 0 (Glycogen Synthase Deficiency) – 112 6.2 The Muscle Glycogenoses – 112 6.2.1 Glycogen Storage Disease Type V (Myophosphorylase Deficiency) – 113 6.2.2 Glycogen Storage Disease Type VII (Phosphofructokinase Deficiency) – 113 6.2.3 Phosphoglycerate Kinase Deficiency – 114 6.2.4 Glycogen Storage Disease Type X (Phosphoglycerate Mutase Deficiency) – 114 6.2.5 Glycogen Storage Disease Type XII (Aldolase A Deficiency) – 114 6.2.6 Glycogen Storage Disease Type XIII (E-Enolase Deficiency) – 115 6.2.7 Glycogen Storage Disease Type XI (Lactate Dehydrogenase Deficiency) – 115 6.2.8 Muscle Glycogen Storage Disease Type 0 (Glycogen Synthase Deficiency) – 115 6.3 The Generalized Glycogenoses and Related Disorders – 115 6.3.1 Glycogen Storage Disease Type II (Acid Maltase Deficiency) – 115 6.3.2 Danon Disease – 116 6.3.3 Lafora Disease – 116 References – 116 102 Chapter 6 · The Glycogen Storage Diseases and Related Disorders Glycogen Metabolism Glycogen is a macromolecule composed of glucose viding glucose and glycolytic intermediates (. Fig. 6.1). units. -

Murine Obscurin and Obsl1 Have Functionally Redundant Roles in Sarcolemmal Integrity, Sarcoplasmic Reticulum Organization, and Muscle Metabolism
UC San Diego UC San Diego Previously Published Works Title Murine obscurin and Obsl1 have functionally redundant roles in sarcolemmal integrity, sarcoplasmic reticulum organization, and muscle metabolism. Permalink https://escholarship.org/uc/item/46t7g5hw Journal Communications biology, 2(1) ISSN 2399-3642 Authors Blondelle, Jordan Marrocco, Valeria Clark, Madison et al. Publication Date 2019 DOI 10.1038/s42003-019-0405-7 Peer reviewed eScholarship.org Powered by the California Digital Library University of California ARTICLE https://doi.org/10.1038/s42003-019-0405-7 OPEN Murine obscurin and Obsl1 have functionally redundant roles in sarcolemmal integrity, sarcoplasmic reticulum organization, and muscle metabolism 1234567890():,; Jordan Blondelle1,7, Valeria Marrocco1,7, Madison Clark1, Patrick Desmond1, Stephanie Myers1, Jim Nguyen1, Matthew Wright1, Shannon Bremner2, Enrico Pierantozzi3, Samuel Ward2, Eric Estève1,4, Vincenzo Sorrentino 3, Majid Ghassemian5 & Stephan Lange 1,6 Biological roles of obscurin and its close homolog Obsl1 (obscurin-like 1) have been enig- matic. While obscurin is highly expressed in striated muscles, Obsl1 is found ubiquitously. Accordingly, obscurin mutations have been linked to myopathies, whereas mutations in Obsl1 result in 3M-growth syndrome. To further study unique and redundant functions of these closely related proteins, we generated and characterized Obsl1 knockouts. Global Obsl1 knockouts are embryonically lethal. In contrast, skeletal muscle-specific Obsl1 knockouts show a benign phenotype similar to obscurin knockouts. Only deletion of both proteins and removal of their functional redundancy revealed their roles for sarcolemmal stability and sarcoplasmic reticulum organization. To gain unbiased insights into changes to the muscle proteome, we analyzed tibialis anterior and soleus muscles by mass spectrometry, unco- vering additional changes to the muscle metabolism. -

Danon Disease
Xu et al. Diagnostic Pathology (2021) 16:39 https://doi.org/10.1186/s13000-021-01100-8 CASE REPORT Open Access Danon disease: a case report and literature review Jiamin Xu1†, Zhu Li1†, Yihai Liu1†, Xinlin Zhang1, Fengnan Niu2, Hongyan Zheng1, Lian Wang1, Lina Kang1* , Kun Wang1* and Biao Xu1* Abstract Background: Danon disease (DD) is a rare x-linked dominant multisystemic disorder with a clinical triad of severe cardiomyopathy, skeletal myopathy, and mental retardation. It is caused by a defect in the lysosomal-associated membrane protein-2 (LAMP2) gene, which leads to the formation of autophagic vacuoles containing glycogen granule deposits in skeletal and cardiac muscle fibers. So far, more than 50 different mutations in LAMP2 have been identified. Case presentation: Here, we report an 18-year-old male patient who was hospitalized for heart failure. Biopsy of the left lateral femoral muscle revealed scattered autophagic vacuoles in the muscle fibers with increased glycogen. Next generation sequencing (NGS) was used to detect gene mutations of the proband sample and a novel frameshift mutation (c.1052delG) has been identified in exon 8 of LAMP2, which leads to truncation of the protein. Conclusion: We found a novel frameshift mutation, a hemizygous mutation (c.1052delG) in exon 8 of LAMP2, identified as presenting the hypertrophic cardiomyopathy (HCM) phenotype. Genetic analysis is the gold standard for the diagnosis of DD and is essential to determine appropriate treatment strategies and to confirm the genetic risk of family members. Keywords: Danon disease, Cardiomyopathy, LAMP2, Mutation, NGS Background maturation. Autophagy is a very important biological DD is a rare x-linked dominant multisystemic disorder phenomenon involved in regulating the metabolic balance with clinical manifestations of severe cardiomyopathy, between the synthesis, degradation and reuse of cellular skeletal myopathy, and mental retardation. -

Cardiovascular Diseases Genetic Testing Program Information
Cardiovascular Diseases Genetic Testing Program Description: Congenital Heart Disease Panels We offer comprehensive gene panels designed to • Congenital Heart Disease Panel (187 genes) diagnose the most common genetic causes of hereditary • Heterotaxy Panel (114 genes) cardiovascular diseases. Testing is available for congenital • RASopathy/Noonan Spectrum Disorders Panel heart malformation, cardiomyopathy, arrythmia, thoracic (31 genes) aortic aneurysm, pulmonary arterial hypertension, Marfan Other Panels syndrome, and RASopathy/Noonan spectrum disorders. • Pulmonary Arterial Hypertension (PAH) Panel Hereditary cardiovascular disease is caused by variants in (20 genes) many different genes, and may be inherited in an autosomal dominant, autosomal recessive, or X-linked manner. Other Indications: than condition-specific panels, we also offer single gene Panels: sequencing for any gene on the panels, targeted variant • Confirmation of genetic diagnosis in a patient with analysis, and targeted deletion/duplication analysis. a clinical diagnosis of cardiovascular disease Tests Offered: • Carrier or pre-symptomatic diagnosis identification Arrythmia Panels in individuals with a family history of cardiovascular • Comprehensive Arrhythmia Panel (81 genes) disease of unknown genetic basis • Atrial Fibrillation (A Fib) Panel (28 genes) Gene Specific Sequencing: • Atrioventricular Block (AV Block) Panel (7 genes) • Confirmation of genetic diagnosis in a patient with • Brugada Syndrome Panel (21 genes) cardiovascular disease and in whom a specific -

Genetic Testing Medical Policy – Genetics
Genetic Testing Medical Policy – Genetics Please complete all appropriate questions fully. Suggested medical record documentation: • Current History & Physical • Progress Notes • Family Genetic History • Genetic Counseling Evaluation *Failure to include suggested medical record documentation may result in delay or possible denial of request. PATIENT INFORMATION Name: Member ID: Group ID: PROCEDURE INFORMATION Genetic Counseling performed: c Yes c No **Please check the requested analyte(s), identify number of units requested, and provide indication/rationale for testing. 81400 Molecular Pathology Level 1 Units _____ c ACADM (acyl-CoA dehydrogenase, C-4 to C-12 straight chain, MCAD) (e.g., medium chain acyl dehydrogenase deficiency), K304E variant _____ c ACE (angiotensin converting enzyme) (e.g., hereditary blood pressure regulation), insertion/deletion variant _____ c AGTR1 (angiotensin II receptor, type 1) (e.g., essential hypertension), 1166A>C variant _____ c BCKDHA (branched chain keto acid dehydrogenase E1, alpha polypeptide) (e.g., maple syrup urine disease, type 1A), Y438N variant _____ c CCR5 (chemokine C-C motif receptor 5) (e.g., HIV resistance), 32-bp deletion mutation/794 825del32 deletion _____ c CLRN1 (clarin 1) (e.g., Usher syndrome, type 3), N48K variant _____ c DPYD (dihydropyrimidine dehydrogenase) (e.g., 5-fluorouracil/5-FU and capecitabine drug metabolism), IVS14+1G>A variant _____ c F13B (coagulation factor XIII, B polypeptide) (e.g., hereditary hypercoagulability), V34L variant _____ c F2 (coagulation factor 2) (e.g., -
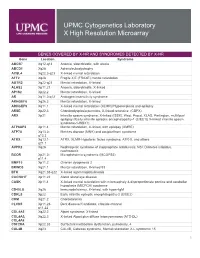
Genes Covered and Disorders Detected by X-HR Microarray
UPMC Cytogenetics Laboratory X High Resolution Microarray GENES COVERED BY X-HR AND SYNDROMES DETECTED BY X-HR Gene Location Syndrome ABCB7 Xq12-q13 Anemia, sideroblastic, with ataxia ABCD1 Xq28 Adrenoleukodystrophy ACSL4 Xq22.3-q23 X-linked mental retardation AFF2 Xq28 Fragile X E (FRAXE) mental retardation AGTR2 Xq22-q23 Mental retardation, X-linked ALAS2 Xp11.21 Anemia, sideroblastic, X-linked AP1S2 Xp22.2 Mental retardation, X-linked AR Xq11.2-q12 Androgen insensitivity syndrome ARHGEF6 Xq26.3 Mental retardation, X-linked ARHGEF9 Xq11.1 X-linked mental retardation (XLMR)//Hyperekplexia and epilepsy ARSE Xp22.3 Chondrodysplasia punctata, X-linked recessive (CDPX) ARX Xp21 Infantile spasm syndrome, X-linked (ISSX), West, Proud, XLAG, Partington, multifocal epilepsy //Early infantile epileptic encephalopathy-1 (EIEE1)( X-linked infantile spasm syndrome-1-ISSX1) ATP6AP2 Xp11.4 Mental retardation, X-linked, with epilepsy (XMRE) ATP7A Xq13.2- Menkes disease (MNK) and occipital horn syndrome q13.3 ATRX Xq13.1- ATRX, XLMR-Hypotonic facies syndrome, ATR-X, and others q21.1 AVPR2 Xq28 Nephrogenic syndrome of inappropriate antidiuresis; NSI; Diabetes insipidus, nephrogenic BCOR Xp21.2- Microphthalmia syndromic (MCOPS2) p11.4 BMP15 Xp11.2 Ovarian dysgenesis 2 BRWD3 Xq21.1 Mental retardation, X-linked 93 BTK Xq21.33-q22 X-linked agammaglobulinemia CACNA1F Xp11.23 Aland Island eye disease CASK Xp11.4 X-linked mental retardation with microcephaly & disproportionate pontine and cerebellar hypoplasia (MICPCH) syndrome CD40LG Xq26 Immunodeficiency, -

Supplementary Table 1: Genes Located on Chromosome 18P11-18Q23, an Area Significantly Linked to TMPRSS2-ERG Fusion
Supplementary Table 1: Genes located on Chromosome 18p11-18q23, an area significantly linked to TMPRSS2-ERG fusion Symbol Cytoband Description LOC260334 18p11 HSA18p11 beta-tubulin 4Q pseudogene IL9RP4 18p11.3 interleukin 9 receptor pseudogene 4 LOC100132166 18p11.32 hypothetical LOC100132166 similar to Rho-associated protein kinase 1 (Rho- associated, coiled-coil-containing protein kinase 1) (p160 LOC727758 18p11.32 ROCK-1) (p160ROCK) (NY-REN-35 antigen) ubiquitin specific peptidase 14 (tRNA-guanine USP14 18p11.32 transglycosylase) THOC1 18p11.32 THO complex 1 COLEC12 18pter-p11.3 collectin sub-family member 12 CETN1 18p11.32 centrin, EF-hand protein, 1 CLUL1 18p11.32 clusterin-like 1 (retinal) C18orf56 18p11.32 chromosome 18 open reading frame 56 TYMS 18p11.32 thymidylate synthetase ENOSF1 18p11.32 enolase superfamily member 1 YES1 18p11.31-p11.21 v-yes-1 Yamaguchi sarcoma viral oncogene homolog 1 LOC645053 18p11.32 similar to BolA-like protein 2 isoform a similar to 26S proteasome non-ATPase regulatory LOC441806 18p11.32 subunit 8 (26S proteasome regulatory subunit S14) (p31) ADCYAP1 18p11 adenylate cyclase activating polypeptide 1 (pituitary) LOC100130247 18p11.32 similar to cytochrome c oxidase subunit VIc LOC100129774 18p11.32 hypothetical LOC100129774 LOC100128360 18p11.32 hypothetical LOC100128360 METTL4 18p11.32 methyltransferase like 4 LOC100128926 18p11.32 hypothetical LOC100128926 NDC80 homolog, kinetochore complex component (S. NDC80 18p11.32 cerevisiae) LOC100130608 18p11.32 hypothetical LOC100130608 structural maintenance -

Generation of Induced Pluripotent Stem Cells from a Female Patient with a Xq27.3-Q28 Deletion to Establish Disease Models and Identify Therapies
CELLULAR REPROGRAMMING Volume 22, Number 4, 2020 Research Article ª Mary Ann Liebert, Inc. DOI: 10.1089/cell.2020.0012 Generation of Induced Pluripotent Stem Cells from a Female Patient with a Xq27.3-q28 Deletion to Establish Disease Models and Identify Therapies Noriko Watanabe,1,* Kohei Kitada,1,* Katherine E. Santostefano,1 Airi Yokoyama,1 Sara M. Waldrop,1 Coy D. Heldermon,2 Daisuke Tachibana,3 Masayasu Koyama,3 Amy M. Meacham,2 Christina A. Pacak,4 and Naohiro Terada1 Abstract Since it is extremely difficult to establish an animal model for human chromosomal abnormalities, induced pluripotent stem cells (iPSCs) provide a powerful alternative to study underlying mechanisms of these disorders and identify potential therapeutic interventions. In this study we established iPSCs from a young girl with a hemizygous deletion of Xq27.3-q28 who exhibited global developmental delay and intellectual disability from early in infancy. The deletion site on the X chromosome includes Fragile X Mental Retardation 1 (FMR1), the gene responsible for fragile X syndrome, which likely contributes to the patient’s neurodevelopmental ab- normalities. The FMR1 gene was expressed in approximately half of the iPSC clones we generated while it was absent in the other half due to the random inactivation of normal and abnormal X chromosomes. The normal or absent expression pattern of the FMR1 gene was not altered when the iPSCs were differentiated into neural progenitor cells (NPCs). Moreover, chromosome reactivating reagents such as 5-aza-2-deoxycytidine, tri- chostatin A, and UNC0638, were tested in an attempt to reactivate the suppressed FMR1 gene in affected iPSC- NPCs.