The Impact of X-Chromosome Inactivation on Phenotypic Expression of X-Linked Neurodevelopmental Disorders
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The University of Chicago Genetic Services Laboratories
The University of Chicago Genetic Services Laboratories 5841 S. Maryland Ave., Rm. G701, MC 0077, Chicago, Illinois 60637 Toll Free: (888) UC GENES (888) 824 3637 Local: (773) 834 0555 FAX: (773) 702 9130 [email protected] dnatesting.uchicago.edu. CLIA #: 14D0917593 CAP #: 18827-49 Next Generation Sequencing Panel for Early Infantile Epileptic Encephalopathy Clinical Features and Molecular Genetics: Early infantile epileptic encephalopathy (EIEE), also known as Ohtahara syndrome, is a severe form of epilepsy characterized by frequent tonic spasms with onset in the first months of life. EEG reveals suppression-burst patterns, characterized by high- voltage bursts alternating with almost flat suppression phases. Seizures are medically intractable with evolution to West syndrome at 3-6 months of age and then Lennox-Gastaut syndrome at 1-3 years of age. EIEE represents approximately 1% of all epilepsies occuring in children less than 15 years of age (1). Patients have severe developmental delay and poor prognosis. The diagnostic workup of EIEEs remains challenging because of frequent difficulties in defining etiologies. Acquired structural abnormalities like hypoxic-ischemic insults and isolated cortical malformations, which represent the most common causes of epileptic encelphalopathy in infancy should be excluded first (2). Our EIEE Panel includes sequence analysis of all 15 genes listed below, and deletion/duplication analysis of the 12 genes listed in bold below. Early Infantile Epileptic Encephalopathy Panel ARHGEF9 KCNQ2 PNKP SLC2A1 ARX MAGI2 SCN1A SPTAN1 CDKL5 PCDH19 SCN2A STXBP1 GRIN2A PLCB1 SLC25A22 Gene / Condition Clinical Features and Molecular Pathology ARHGEF9 The ARHGEF9 gene encodes the protein collybistin, which is a brain-specific protein involved in EIEE8 inhibitory synaptogenesis (3). -

Childhood Cerebral X-Linked Adrenoleukodystrophy with Atypical Neuroimaging Abnormalities and a No…
9/28/2018 Journal of Postgraduate Medicine: Childhood cerebral X-linked adrenoleukodystrophy with atypical neuroimaging abnormalities and a no… Open access journal indexed with Index Medicus & EMBASE Home | Subscribe | Feedback [Download PDF] CASE REPORT Year : 2018 | Volume : 64 | Issue : 1 | Page : 59-63 Childhood cerebral X-linked adrenoleukodystrophy with atypical neuroimaging abnormalities and a novel mutation M Muranjan1, S Karande1, S Sankhe2, S Eichler3, 1 Department of Pediatrics, Seth GS Medical College and KEM Hospital, Parel, Mumbai, Maharashtra, India 2 Department of Radiology, Seth GS Medical College and KEM Hospital, Parel, Mumbai, Maharashtra, India 3 Centogene AG, Schillingallee 68, Rostock, Germany Correspondence Address: Dr. M Muranjan Department of Pediatrics, Seth GS Medical College and KEM Hospital, Parel, Mumbai, Maharashtra India Abstract Childhood cerebral X-linked adrenoleukodystrophy (XALD) typically manifests with symptoms of adrenocortical insufficiency and a variety of neurocognitive and behavioral abnormalities. A major diagnostic clue is the characteristic neuroinflammatory parieto-occipital white matter lesions on magnetic resonance imaging. This study reports a 5-year 10-month old boy presenting with generalized skin hyperpigmentation since 3 years of age. Over the past 9 months, he had developed right-sided hemiparesis and speech and behavioral abnormalities, which had progressed over 5 months to bilateral hemiparesis. Retrospective analyses of serial brain magnetic resonance images revealed an unusual pattern of lesions involving the internal capsules, corticospinal tracts in the midbrain and brainstem, and cerebellar white matter. The clinical diagnosis of childhood cerebral adrenoleukodystrophy was confirmed by elevated basal levels of adrenocorticotropin hormone and plasma very long chain fatty acid levels. Additionally, sequencing of the ABCD1 gene revealed a novel mutation. -

Coupling of Spliceosome Complexity to Intron Diversity
bioRxiv preprint doi: https://doi.org/10.1101/2021.03.19.436190; this version posted March 20, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Coupling of spliceosome complexity to intron diversity Jade Sales-Lee1, Daniela S. Perry1, Bradley A. Bowser2, Jolene K. Diedrich3, Beiduo Rao1, Irene Beusch1, John R. Yates III3, Scott W. Roy4,6, and Hiten D. Madhani1,6,7 1Dept. of Biochemistry and Biophysics University of California – San Francisco San Francisco, CA 94158 2Dept. of Molecular and Cellular Biology University of California - Merced Merced, CA 95343 3Department of Molecular Medicine The Scripps Research Institute, La Jolla, CA 92037 4Dept. of Biology San Francisco State University San Francisco, CA 94132 5Chan-Zuckerberg Biohub San Francisco, CA 94158 6Corresponding authors: [email protected], [email protected] 7Lead Contact 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.03.19.436190; this version posted March 20, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. SUMMARY We determined that over 40 spliceosomal proteins are conserved between many fungal species and humans but were lost during the evolution of S. cerevisiae, an intron-poor yeast with unusually rigid splicing signals. We analyzed null mutations in a subset of these factors, most of which had not been investigated previously, in the intron-rich yeast Cryptococcus neoformans. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

The Inactive X Chromosome Is Epigenetically Unstable and Transcriptionally Labile in Breast Cancer
Supplemental Information The inactive X chromosome is epigenetically unstable and transcriptionally labile in breast cancer Ronan Chaligné1,2,3,8, Tatiana Popova1,4, Marco-Antonio Mendoza-Parra5, Mohamed-Ashick M. Saleem5 , David Gentien1,6, Kristen Ban1,2,3,8, Tristan Piolot1,7, Olivier Leroy1,7, Odette Mariani6, Hinrich Gronemeyer*5, Anne Vincent-Salomon*1,4,6,8, Marc-Henri Stern*1,4,6 and Edith Heard*1,2,3,8 Extended Experimental Procedures Cell Culture Human Mammary Epithelial Cells (HMEC, Invitrogen) were grown in serum-free medium (HuMEC, Invitrogen). WI- 38, ZR-75-1, SK-BR-3 and MDA-MB-436 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen) containing 10% fetal bovine serum (FBS). DNA Methylation analysis. We bisulfite-treated 2 µg of genomic DNA using Epitect bisulfite kit (Qiagen). Bisulfite converted DNA was amplified with bisulfite primers listed in Table S3. All primers incorporated a T7 promoter tag, and PCR conditions are available upon request. We analyzed PCR products by MALDI-TOF mass spectrometry after in vitro transcription and specific cleavage (EpiTYPER by Sequenom®). For each amplicon, we analyzed two independent DNA samples and several CG sites in the CpG Island. Design of primers and selection of best promoter region to assess (approx. 500 bp) were done by a combination of UCSC Genome Browser (http://genome.ucsc.edu) and MethPrimer (http://www.urogene.org). All the primers used are listed (Table S3). NB: MAGEC2 CpG analysis have been done with a combination of two CpG island identified in the gene core. Analysis of RNA allelic expression profiles (based on Human SNP Array 6.0) DNA and RNA hybridizations were normalized by Genotyping console. -

Neurodegenerative Triplet Repeat Expansion Disorders
The Pharma Innovation Journal 2018; 7(11): 34-40 ISSN (E): 2277- 7695 ISSN (P): 2349-8242 NAAS Rating: 5.03 Neurodegenerative triplet repeat expansion disorders: TPI 2018; 7(11): 34-40 © 2018 TPI A review www.thepharmajournal.com Received: 15-09-2018 Accepted: 20-10-2018 Mitesh Patel, RK Patel, Tushar Chauhan, Jigar Suthar and Sanjay Dave Mitesh Patel Genetics Group of Gujarat Abstract Diagnostic Centre, Mehsana, Epigenetic alterations are the major causes of triplet repeat expansion. The repetitive DNA expands of its Gujarat, India normal length results in sever neurodegenerative conditions. The common types of triplet repeat expansion (TNE) disorders are: Huntington disease, Friedreich ataxia, myotonic dystrophy, SBMA and RK Patel SCA1 out of which Huntington disease, SBMA and SCA1 are categorized as a poly glutamine disorder Sandor Animal Biogenics Pvt. due to the repeat of CAG. In contrast, the friedreich ataxia is occurred due to expansion of the GAA Ltd., Hyderabad, Telangana, whereas myotonic dystrophy is due to the expansion of CTG. The triplet disease follows the mechanism India of anticipation in which the onset of the disease increases with age. Conclusively, no clear mechanism can explain the origin of the disease. The pre mutation can be expanded in full mutation in successive Tushar Chauhan Genetics Group of Gujarat generations and the number of repeats increased with each generation. TNE can observe in both somatic Diagnostic Centre, Mehsana, as well as germ line tissues. Gujarat, India Keywords: triplet repeat expansion disorder, trinucleotide repeats, Huntington disease, SBMA, SCA1, Jigar Suthar friedreich ataxia Genetics Group of Gujarat Diagnostic Centre, Mehsana, Introduction Gujarat, India Since the early 1990s, a new class of molecular disease has been characterized based upon the Sanjay Dave presence of unstable and abnormal expansions of DNA-triplets (Trinucleotides). -

Primate Specific Retrotransposons, Svas, in the Evolution of Networks That Alter Brain Function
Title: Primate specific retrotransposons, SVAs, in the evolution of networks that alter brain function. Olga Vasieva1*, Sultan Cetiner1, Abigail Savage2, Gerald G. Schumann3, Vivien J Bubb2, John P Quinn2*, 1 Institute of Integrative Biology, University of Liverpool, Liverpool, L69 7ZB, U.K 2 Department of Molecular and Clinical Pharmacology, Institute of Translational Medicine, The University of Liverpool, Liverpool L69 3BX, UK 3 Division of Medical Biotechnology, Paul-Ehrlich-Institut, Langen, D-63225 Germany *. Corresponding author Olga Vasieva: Institute of Integrative Biology, Department of Comparative genomics, University of Liverpool, Liverpool, L69 7ZB, [email protected] ; Tel: (+44) 151 795 4456; FAX:(+44) 151 795 4406 John Quinn: Department of Molecular and Clinical Pharmacology, Institute of Translational Medicine, The University of Liverpool, Liverpool L69 3BX, UK, [email protected]; Tel: (+44) 151 794 5498. Key words: SVA, trans-mobilisation, behaviour, brain, evolution, psychiatric disorders 1 Abstract The hominid-specific non-LTR retrotransposon termed SINE–VNTR–Alu (SVA) is the youngest of the transposable elements in the human genome. The propagation of the most ancient SVA type A took place about 13.5 Myrs ago, and the youngest SVA types appeared in the human genome after the chimpanzee divergence. Functional enrichment analysis of genes associated with SVA insertions demonstrated their strong link to multiple ontological categories attributed to brain function and the disorders. SVA types that expanded their presence in the human genome at different stages of hominoid life history were also associated with progressively evolving behavioural features that indicated a potential impact of SVA propagation on a cognitive ability of a modern human. -

Genetic and Neurodevelopmental Spectrum Of
Cognitive and behavioural genetics J Med Genet: first published as 10.1136/jmedgenet-2015-103451 on 17 March 2016. Downloaded from ORIGINAL ARTICLE Genetic and neurodevelopmental spectrum of SYNGAP1-associated intellectual disability and epilepsy Cyril Mignot,1,2,3 Celina von Stülpnagel,4,5 Caroline Nava,1,6 Dorothée Ville,7 Damien Sanlaville,8,9,10 Gaetan Lesca,8,9,10 Agnès Rastetter,6 Benoit Gachet,6 Yannick Marie,6 G Christoph Korenke,11 Ingo Borggraefe,12 Dorota Hoffmann-Zacharska,13 Elżbieta Szczepanik,14 Mariola Rudzka-Dybała,14 Uluç Yiş,15 Hande Çağlayan,16 Arnaud Isapof,17 Isabelle Marey,1 Eleni Panagiotakaki,18 Christian Korff,19 Eva Rossier,20 Angelika Riess,21 Stefanie Beck-Woedl,21 Anita Rauch,22 Christiane Zweier,23 Juliane Hoyer,23 André Reis,23 Mikhail Mironov,24 Maria Bobylova,24 Konstantin Mukhin,24 Laura Hernandez-Hernandez,25 Bridget Maher,25 Sanjay Sisodiya,25 Marius Kuhn,26 Dieter Glaeser,26 Sarah Weckhuysen,6,27 Candace T Myers,28 Heather C Mefford,28 Konstanze Hörtnagel,29 Saskia Biskup,29 EuroEPINOMICS-RES MAE working group, Johannes R Lemke,30 Delphine Héron,1,2,3,4 Gerhard Kluger,4,5 Christel Depienne1,6 ▸ Additional material is ABSTRACT INTRODUCTION published online only. To view Objective We aimed to delineate the neurodevelopmental The human SYNGAP1 gene on chromosome please visit the journal online (http://dx.doi.org/10.1136/ spectrum associated with SYNGAP1 mutations and to 6p21.3 encodes the synaptic RAS-GTPase-activating jmedgenet-2015-103451). investigate genotype–phenotype correlations. protein 1, a protein of the post-synaptic density Methods We sequenced the exome or screened the exons (PSD) of glutamatergic neurons.12SYNGAP1 inter- For numbered affiliations see end of article. -
![Downloaded from [266]](https://docslib.b-cdn.net/cover/7352/downloaded-from-266-347352.webp)
Downloaded from [266]
Patterns of DNA methylation on the human X chromosome and use in analyzing X-chromosome inactivation by Allison Marie Cotton B.Sc., The University of Guelph, 2005 A THESIS SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY in The Faculty of Graduate Studies (Medical Genetics) THE UNIVERSITY OF BRITISH COLUMBIA (Vancouver) January 2012 © Allison Marie Cotton, 2012 Abstract The process of X-chromosome inactivation achieves dosage compensation between mammalian males and females. In females one X chromosome is transcriptionally silenced through a variety of epigenetic modifications including DNA methylation. Most X-linked genes are subject to X-chromosome inactivation and only expressed from the active X chromosome. On the inactive X chromosome, the CpG island promoters of genes subject to X-chromosome inactivation are methylated in their promoter regions, while genes which escape from X- chromosome inactivation have unmethylated CpG island promoters on both the active and inactive X chromosomes. The first objective of this thesis was to determine if the DNA methylation of CpG island promoters could be used to accurately predict X chromosome inactivation status. The second objective was to use DNA methylation to predict X-chromosome inactivation status in a variety of tissues. A comparison of blood, muscle, kidney and neural tissues revealed tissue-specific X-chromosome inactivation, in which 12% of genes escaped from X-chromosome inactivation in some, but not all, tissues. X-linked DNA methylation analysis of placental tissues predicted four times higher escape from X-chromosome inactivation than in any other tissue. Despite the hypomethylation of repetitive elements on both the X chromosome and the autosomes, no changes were detected in the frequency or intensity of placental Cot-1 holes. -

Centre for Arab Genomic Studies a Division of Sheikh Hamdan Award for Medical Sciences
Centre for Arab Genomic Studies A Division of Sheikh Hamdan Award for Medical Sciences The Catalogue for Transmission Genetics in Arabs CTGA Database tRNA Methyltransferase 1, S. Cerevisiae, Homolog of Alternative Names planus. These findings are further bolstered by the TRMT1 fact that mutations in two other RNA- N2,N2-Dimethylguanosine-26 tRNA methyltransferase genes, NSUN2 and FTSJ1, have Methyltransferase been associated with intellectual disability. tRNA(m(2,2)G26)Dimethyltransferase Record Category Molecular Genetics Gene locus The TRMT1 gene is located on the short arm of chromosome 19 and spans a length of 12.6 kb of WHO-ICD DNA. Its coding sequence is spread across 18 N/A to gene loci exons and it encodes a 72.2 kDa protein product comprised of 659 amino acids. An additional 69.3 Incidence per 100,000 Live Births kDa isoform of the TRMT1 protein exists due to an N/A to gene loci alternatively spliced transcript variant. The gene is widely expressed in the human body, particularly in OMIM Number the nervous system, intestine, spleen, kidney and 611669 lung. Mode of Inheritance Epidemiology in the Arab World N/A to gene loci Saudi Arabia Monies et al. (2017) studied the findings of 1000 Gene Map Locus diagnostic panels and exomes carried out at a next 19p13.13 generation sequencing lab in Saudi Arabia. One patient, a 13-year-old male, presented with speech Description delay, intellectual disability, learning disability, The TRMT1 gene encodes a methyltransferase hypotonia and seizures. Using whole exome enzyme that acts on tRNA. This enzyme, which sequencing, a homozygous mutation consists of a zinc finger motif and an (c.1245_1246del, p.L415fs) was identified in exon arginine/proline rich region at its C-terminus, is 10 of the patient’s TRMT1 gene. -

Nicole Trask, Pharmd, Planning for the 2019 Specialty Drug Spend
Planning for the 2019 Specialty Drug Spend August 24, 2018 Nicole Trask, PharmD Clinical Consultant Pharmacist University of Massachusetts – Clinical Pharmacy Services Disclosure for Nicole Trask I have no actual or potential conflict of interest in relation to this presentation. Budget Impact Modeling for 2 ||August 24, 2018 the Specialty Drug Spend Objectives • Identify high-impact specialty pipeline drugs expected to reach the market in 2019-2020 • Summarize efficacy data for high-impact specialty pipeline drugs and indicate their anticipated place in therapy • Compare specialty pipeline drugs to currently available therapeutic options • Predict the budgetary impact of specialty pipeline drugs and discuss strategies to mitigate costs Budget Impact Modeling for 3 ||August 24, 2018 the Specialty Drug Spend Identifying High-Impact Drugs Two key drivers • Clinical impact • Efficacy/effectiveness • Therapeutic alternatives • Economic impact • Cost • Volume Budget Impact Modeling for 4 ||August 24, 2018 the Specialty Drug Spend Assessing Clinical Impact Clinical trial data Therapeutic alternatives • Placebo-controlled, • Me-too drug vs. head-to-head studies first-in-class • Adverse events • Market competition • Potential drug-drug • Consensus interactions guidelines • Target population • Patient willingness to use medication Budget Impact Modeling for 5 ||August 24, 2018 the Specialty Drug Spend Assessing Economic Impact Cost Volume • NADAC, AWP, WAC • Prevalence/incidence of • Supplemental rebate disease • Outcomes-based • Frequency of contracts administration • Value assessments • Duration of therapy (e.g., AHRQ, ICER, PCORI) AHRQ=Agency for Healthcare Research and Quality, AWP=average wholesale price, ICER=Institute for Clinical and Economic Review, NADAC=national average drug acquisition cost, PCORI=Patient-centered Outcomes Research Institute, WAC=wholesale acquisition cost Budget Impact Modeling for 6 ||August 24, 2018 the Specialty Drug Spend Other Factors Affecting Budget Impact Disease-specific Prescriber-specific • Chronic vs. -
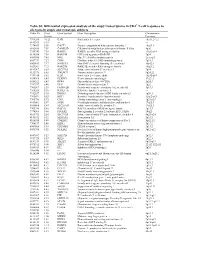
Table S1| Differential Expression Analysis of the Atopy Transcriptome
Table S1| Differential expression analysis of the atopy transcriptome in CD4+ T-cell responses to allergens in atopic and nonatopic subjects Probe ID S.test Gene Symbol Gene Description Chromosome Statistic Location 7994280 10.32 IL4R Interleukin 4 receptor 16p11.2-12.1 8143383 8.95 --- --- --- 7974689 8.50 DACT1 Dapper, antagonist of beta-catenin, homolog 1 14q23.1 8102415 7.59 CAMK2D Calcium/calmodulin-dependent protein kinase II delta 4q26 7950743 7.58 RAB30 RAB30, member RAS oncogene family 11q12-q14 8136580 7.54 RAB19B GTP-binding protein RAB19B 7q34 8043504 7.45 MAL Mal, T-cell differentiation protein 2cen-q13 8087739 7.27 CISH Cytokine inducible SH2-containing protein 3p21.3 8000413 7.17 NSMCE1 Non-SMC element 1 homolog (S. cerevisiae) 16p12.1 8021301 7.15 RAB27B RAB27B, member RAS oncogene family 18q21.2 8143367 6.83 SLC37A3 Solute carrier family 37 member 3 7q34 8152976 6.65 TMEM71 Transmembrane protein 71 8q24.22 7931914 6.56 IL2R Interleukin 2 receptor, alpha 10p15-p14 8014768 6.43 PLXDC1 Plexin domain containing 1 17q21.1 8056222 6.43 DPP4 Dipeptidyl-peptidase 4 (CD26) 2q24.3 7917697 6.40 GFI1 Growth factor independent 1 1p22 7903507 6.39 FAM102B Family with sequence similarity 102, member B 1p13.3 7968236 5.96 RASL11A RAS-like, family 11, member A --- 7912537 5.95 DHRS3 Dehydrogenase/reductase (SDR family) member 3 1p36.1 7963491 5.83 KRT1 Keratin 1 (epidermolytic hyperkeratosis) 12q12-q13 7903786 5.72 CSF1 Colony stimulating factor 1 (macrophage) 1p21-p13 8019061 5.67 SGSH N-sulfoglucosamine sulfohydrolase (sulfamidase) 17q25.3