Alkenes and Alkynes and Rad on React
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Investigation of Base-Free Copper-Catalysed Azide–Alkyne Click Cycloadditions (Cuaac) in Natural Deep Eutectic Solvents As Green and Catalytic Reaction Media
Investigation of Base-free Copper-Catalysed Azide–Alkyne Click Cycloadditions (CuAAc) in Natural Deep Eutectic Solvents as Green and Catalytic Reaction Media Salvatore V. Giofrè,1* Matteo Tiecco,2* Angelo Ferlazzo,3 Roberto Romeo,1 Gianluca Ciancaleoni,4 Raimondo Germani2 and Daniela Iannazzo3 1. Dipartimento di Scienze Chimiche, Biologiche, Farmaceutiche ed Ambientali, Università di Messina, Viale Annunziata, I-98168 Messina, Italy. 2. Dipartimento di Chimica, Biologia e Biotecnologie, Università di Perugia, via Elce di Sotto 8, I- 06123 Perugia, Italy. 3. Dipartimento di Ingegneria, Università of Messina, Contrada Di Dio, I-98166 Messina, Italy 4. Dipartimento di Chimica e Chimica Industriale (DCCI), Università di Pisa, Via Giuseppe Moruzzi, 13, I-56124 Pisa, Italy. * Corresponding authors Email addresses: [email protected] (Salvatore V. Giofrè); [email protected] (Matteo Tiecco). ABSTRACT The click cycloaddition reaction of azides and alkynes affording 1,2,3-triazoles is a transformation widely used to obtain relevant products in chemical biology, medicinal chemistry, materials science and other fields. In this work, a set of Natural Deep Eutectic Solvents (NADESs) as “active” reaction media has been investigated in the copper-catalysed azide–alkyne cycloaddition reactions (CuAAc). The use of these green liquids as green and catalytic solvents has shown to improve the reaction effectiveness, giving excellent yields. The NADESs proved to be “active” in this transformation for the absence of added bases in all the performed reactions and in several cases for their reducing capabilities. The results were rationalized by DFT calculations which demonstrated the involvement of H-bonds between DESs and alkynes as well as a stabilization of copper catalytic intermediates. -

Oligomeric A2 + B3 Approach to Branched Poly(Arylene Ether Sulfone)
“One-Pot” Oligomeric A 2 + B 3 Approach to Branched Poly(arylene ether sulfone)s: Reactivity Ratio Controlled Polycondensation A thesis submitted in partial fulfillment of the requirements for the degree of Master of Science By ANDREA M. ELSEN B.S., Wright State University, 2007 2009 Wright State University WRIGHT STATE UNIVERSITY SCHOOL OF GRADUATE STUDIES June 19, 200 9 I HEREBY RECOMMEND THAT THE THESIS PREPARED UNDER MY SUPERVISION BY Andrea M. Elsen ENTITLED “One-Pot” Oligomeric A2 + B3 Approach to Branched Poly(arylene ether sulfone)s: Reactivity Ratio Controlled Polycondenstation BE ACCEPTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF Master of Science . _________________________ Eric Fossum, Ph.D. Thesis Director _________________________ Kenneth Turnbull, Ph.D. Department Chair Committee on Final Examination ____________________________ Eric Fossum, Ph.D. ____________________________ Kenneth Turnbull, Ph.D. ____________________________ William A. Feld, Ph.D. ____________________________ Joseph F. Thomas, Jr., Ph.D. Dean, School of Graduate Studies Abstract Elsen, Andrea M. M.S., Department of Chemistry, Wright State University, 2009. “One-Pot” Oligomeric A 2 + B 3 Approach to Branched Poly(arylene ether sulfone)s: Reactivity Ratio Controlled Polycondensation The synthesis of fully soluble branched poly(arylene ether)s via an oligomeric A 2 + B 3 system, in which the A 2 oligomers are generated in situ, is presented. This approach takes advantage of the significantly higher reactivity toward nucleophilic aromatic substitution reactions, NAS, of B 2, 4-Fluorophenyl sulfone, relative to B 3, tris (4-Fluorophenyl) phosphine oxide. The A 2 oligomers were synthesized by reaction of Bisphenol-A and B 2, in the presence of the B 3 unit, at temperatures between 100 and 160 °C, followed by an increase in the reaction temperature to 180 °C at which point the branching unit was incorporated. -

Cationic/Anionic/Living Polymerizationspolymerizations Objectives
Chemical Engineering 160/260 Polymer Science and Engineering LectureLecture 1919 -- Cationic/Anionic/LivingCationic/Anionic/Living PolymerizationsPolymerizations Objectives • To compare and contrast cationic and anionic mechanisms for polymerization, with reference to free radical polymerization as the most common route to high polymer. • To emphasize the importance of stabilization of the charged reactive center on the growing chain. • To develop expressions for the average degree of polymerization and molecular weight distribution for anionic polymerization. • To introduce the concept of a “living” polymerization. • To emphasize the utility of anionic and living polymerizations in the synthesis of block copolymers. Effect of Substituents on Chain Mechanism Monomer Radical Anionic Cationic Hetero. Ethylene + - + + Propylene - - - + 1-Butene - - - + Isobutene - - + - 1,3-Butadiene + + - + Isoprene + + - + Styrene + + + + Vinyl chloride + - - + Acrylonitrile + + - + Methacrylate + + - + esters • Almost all substituents allow resonance delocalization. • Electron-withdrawing substituents lead to anionic mechanism. • Electron-donating substituents lead to cationic mechanism. Overview of Ionic Polymerization: Selectivity • Ionic polymerizations are more selective than radical processes due to strict requirements for stabilization of ionic propagating species. Cationic: limited to monomers with electron- donating groups R1 | RO- _ CH =CH- CH =C 2 2 | R2 Anionic: limited to monomers with electron- withdrawing groups O O || || _ -C≡N -C-OR -C- Overview of Ionic Chain Polymerization: Counterions • A counterion is present in both anionic and cationic polymerizations, yielding ion pairs, not free ions. Cationic:~~~C+(X-) Anionic: ~~~C-(M+) • There will be similar effects of counterion and solvent on the rate, stereochemistry, and copolymerization for both cationic and anionic polymerization. • Formation of relatively stable ions is necessary in order to have reasonable lifetimes for propagation. -

Photopolymerization Reactions Under Visible Lights: Principle, Mechanisms and Examples of Applications J.P
Progress in Organic Coatings 47 (2003) 16–36 Photopolymerization reactions under visible lights: principle, mechanisms and examples of applications J.P. Fouassier∗, X. Allonas, D. Burget Département de Photochimie Générale, UMR n◦7525, Ecole Nationale Supérieure de Chimie, 3 Rue Alfred Werner, 68093 Mulhouse Cedex, France Received 22 June 2002; received in revised form 19 November 2002; accepted 20 December 2002 Abstract A general overview of visible light photoinduced polymerization reactions is presented. Reaction mechanisms as well as practical efficiency in industrial applications are discussed. Several points are investigated in detail: photochemical reactivity of photoinitiating system (PIS), short overview of available photoinitiators (PIs) and photosensitizers (PSs), mechanisms involved in selected examples of dye sensitized polymerization reactions, examples of applications in pigmented coatings usable as paints, textile printing, glass reinforced fibers, sunlight curing of waterborne latex paints, curing of inks, laser-induced polymerization reactions, high speed photopolymers for laser imaging, PISs for computer-to-plate systems. © 2003 Elsevier Science B.V. All rights reserved. Keywords: Photopolymerization; Visible light; Radiation curing 1. Introduction Radical, cationic and anionic polymerizations can be ini- tiated by the excitation of suitable photoinitiating systems Radiation curing technologies provide a number of eco- (PISs) under lights. Most of these systems were originally nomic advantages over the usual thermal operation among sensitive to UV lights but by now a large number of various them: rapid through cure, low energy requirements, room systems allows to extend the spectral sensitivity to visible temperature treatment, non-polluting and solvent-free for- lights. According to the applications which are developed, mulations, low costs. -

New Methodologies for the Synthesis Of
NEW METHODOLOGIES FOR THE SYNTHESIS OF ORGANOPHOSPHORUS COMPOUNDS by MONIKA I. ANTCZAK Master of Science, 2002 Wroclaw University of Technology Wroclaw, Poland Submitted to the Graduate Faculty of the College of Science and Engineering Texas Christian University in partial fulfillment of the requirements for the degree of Doctor of Philosophy December 2008 NEW METHODOLOGIESFOR THE SYNTHESISOF ORGANOPHOSPHORUSCOMPOLNDS by MonikaI. Antczak Dissertationapproved: Maior Professor f Scienceand Engineering Copyright by Monika I. Antczak 2008 ACKNOWLEDGMENTS I wish to acknowledge Prof. Jean-Luc Montchamp for the constant interest in the progress of my work over the past five years. I also wish to thank him for his advice and intense discussions, which helped me in expanding my knowledge and understanding of organic chemistry, and in guiding me in my educational progress at Texas Christian University. His patience and support helped me overcome some difficult situation and successfully completing my Ph.D. program. I express my gratitude to my colleagues and friends, Jennifer Tellez, Dr. Laëtitia Coudray, Yamina Belabassi and Dr. Clemence Queffelec for their help, guidance and support. I also would like to thank Dr. Onofrio Annunziata, Dr. Waldek Zerda and Dr. Dale Huckaby for their guidance and interesting discussions during these years. I wish to thank: Monika Wieligor and Dr. Mauricio Quiroz for their friendship and constant support. Finally, I wish to thank my parents for their unconditional love and support. The Chemistry Department at Texas Christian University, the National Science Foundation (CHE-0242898), the National Institute of General Medical Sciences/NIH (R01 GM067610), and the Robert A. Welch Foundation (P-1666) are gratefully acknowledged for the financial support of this research. -

Exam 1 (February 23, 2004) ID# ______
Chemistry 211 Name ___________________________ Exam 1 (February 23, 2004) ID# ___________________________ 10 1. You desire to synthesize 3-ethyl-3-pentanol starting with an ester. (i) What would be the name of the ester, and what is the name for the Grignard reagent (e.g., methyl magnesium bromide)? (ii) For the carbons shown in the product, show plausible hydrocarbons that you could start with to produce the ester and the Grignard reagent (as in a retrosynthesis). 12 2. (i) Show the step-by-step process required to produce propyllithium, which requires a free radical reaction mechanism, . (ii) Show the complete reaction mechanism for reaction between propyllithium and the correct ketone to produce 3-propyl-3-pentanol. (iii) Propose a possible reaction mechanism by which dipropyl cuprate (Cu+ with two propyl groups attached) could react with ethyl bromide to produce a new hydrocarbon. (This is a thinking exercise! So, think! () 8 3. As mentioned in the text, diethyl ether, pentane, and 1-butanol have similar molar masses, but different physical properties. Boiling points are 35oC, 36oC, and 117oC, respectively. Their respective solubilities in water are 7.5g/100mL, insoluble, and 9g/100mL. (i) Draw structures for each of these compounds. (ii) Justify the observed boiling points and their solubilities. 16 4. Draw structures of the following compounds 2,3-heptanediol isopropyllithium benzylmagnesium bromide benzoic acid benzaldehylde dimethyl sulfide t-butyl methanoate dibutyl ketone 12 5. Alcohols can be oxidized to produce other compounds, and can be produced by reduction. For the reactions shown below, show the structure for the expected product (if reaction does not occur, state: No Reaction) when treated with the indicated oxidizing or reducing agents. -
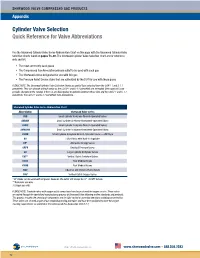
Cylinder Valve Selection Quick Reference for Valve Abbreviations
SHERWOOD VALVE COMPRESSED GAS PRODUCTS Appendix Cylinder Valve Selection Quick Reference for Valve Abbreviations Use the Sherwood Cylinder Valve Series Abbreviation Chart on this page with the Sherwood Cylinder Valve Selection Charts found on pages 73–80. The Sherwood Cylinder Valve Selection Chart are for reference only and list: • The most commonly used gases • The Compressed Gas Association primary outlet to be used with each gas • The Sherwood valves designated for use with this gas • The Pressure Relief Device styles that are authorized by the DOT for use with these gases PLEASE NOTE: The Sherwood Cylinder Valve Selection Charts are partial lists extracted from the CGA V-1 and S-1.1 pamphlets. They can change without notice as the CGA V-1 and S-1.1 pamphlets are amended. Sherwood will issue periodic changes to the catalog. If there is any discrepancy or question between these lists and the CGA V-1 and S-1.1 pamphlets, the CGA V-1 and S-1.1 pamphlets take precedence. Sherwood Cylinder Valve Series Abbreviation Chart Abbreviation Sherwood Valve Series AVB Small Cylinder Acetylene Wrench-Operated Valves AVBHW Small Cylinder Acetylene Handwheel-Operated Valves AVMC Small Cylinder Acetylene Wrench-Operated Valves AVMCHW Small Cylinder Acetylene Handwheel-Operated Valves AVWB Small Cylinder Acetylene Wrench-Operated Valves — WB Style BV Hi/Lo Valves with Built-in Regulator DF* Alternative Energy Valves GRPV Residual Pressure Valves GV Large Cylinder Acetylene Valves GVT** Vertical Outlet Acetylene Valves KVAB Post Medical Valves KVMB Post Medical Valves NGV Industrial and Chrome-Plated Valves YVB† Vertical Outlet Oxygen Valves 1 * DF Valves can be used with all gases; however, the outlet will always be ⁄4"–18 NPT female. -

A Conceptual Design of Fluorous Surfactants Based on a Heterocyclic Core, Put Into Practice Through Synthesis of Fluorous 1,2,3-Triazoles Roger W
Chiang Mai J. Sci. 2009; 36(2) 247 Chiang Mai J. Sci. 2009; 36(2) : 247-257 www.science.cmu.ac.th/journal-science/josci.html Invited Paper A Conceptual Design of Fluorous Surfactants Based on a Heterocyclic Core, Put into Practice Through Synthesis of Fluorous 1,2,3-Triazoles Roger W. Read* and Xiao Bei Wang School of Chemistry, The University of New South Wales, UNSW Sydney NSW 2052, Australia. *Author for correspondence; e-mail: [email protected] ABSTRACT The concept of a design of fluorous surfactants based on a heterocyclic core molecule is introduced and illustrated through the synthesis of polyfluoroalkyl-substituted 1,2,3-triazoles. A two step, one-pot process is described in which a small range of perfluoroalkylethyl azides is prepared in situ and made to undergo Huisgen 1,3-dipolar cycloaddition with terminal alkynes. Measurement of the changes in surface tension of the library of fluorous triazoles at different concentrations in m-xylene reveals unusual behaviour in molecules with a secondary, C8 alkyl substituent. Keywords: organic synthesis, heterocycles, fluorous chemistry, surfactants, click chemistry, polyfluoroalkyl-1,2,3-triazoles. 1. INTRODUCTION Fluorous surfactants have been studied might have applications in the design of new widely, not least for their interest in drug delivery systems and, through self- combination with perfluorocarbons, as assembly, unique molecular devices. emergency blood replacements.[1] Highly Considerable literature exists in the area of fluorinated amphiphiles and colloid systems fluorous block polymers with localised are chemically stable and can carry high crystalline segments and fluorous liposomes concentrations of oxygen and carbon dioxide, and vesicles for drug delivery, but, with a few and therefore have many applications in the exceptions [8-11], most fluorous components biomedical field [2,3]. -

Chemistry for S5 Students Short Notes & Questions with Answers & And
Chemistry for S5 students Short notes & questions with answers & and other questions to help S5 Students to revise Important Concepts: Chemical Reactions of Alkyl Halides The reaction can be broadly classified in two categories: (a) Nucleophilic substitution (b) Elimination reactions Nucleophilic substitution reactions: In this reaction a nucleophile, which is rich in electrons, attacks partial positive charge on the carbon atom bonded to halogen to replace the leaving group. Nucleophilic reactions proceed by two different mechanism: (a) Substitution nucleophilic bimolecular (SN2) (b) Substitution nucleophilic unirnolecular (SN1) The reaction follows second order kinetics No intermediate is formed. It usually requires a strong nucleophile. The order of reactivity followed as: Primary halide > Secondary halide > Tertiary halide It is carried out in polar protic solvents (water, Alcohol, acetic acid etc.). These reactions occur in two steps as shown above The order of leaving ability is: F- < Cl- < Br- < I- The order of reactivity is as shown below: Difference between E1 and E2 reaction mechanism: Attributes E1 E2 Rate law Depend on the concentration of Depends on the concentration of both substrate substrate and base Barrier Formation of carbocation None 3o>2o>> 1o Base Does not require strong base Requires strong base Stereochemistry Does not require stereochemistry Leaving group must be anti to hydrogen removed Some solved questions are given below: Question 1:Which is the correct increasing order of boiling points of the following compounds? 1-bromoethane, 1-bromopropane, 1-bromobutane, Bromobenzene (a) Bromobenzene < 1-bromobutane < 1-bromopropane < 1-bromoethane (b) Bromobenzene < 1-bromothane < 1-bromopropane < 1-bromobutane (c) 1-bromopropane < 1-bromobutane < 1-bromoethane < Bromobenzene (d) 1-bromoethane < 1-bromopropane < 1-bromobutane < Bromobenzene Solution 1: Boiling point increases with increase in size of hydrocarbon part for the same haloalkanes. -

The Chemistry of Radical Polymerization
THE CHEMISTRY OF RADICAL POLYMERIZATION THE CHEMISTRY OF RADICAL POLYMERIZATION GRAEME MOAD CSIRO Molecular and Health Technologies Bayview Ave, Clayton, Victoria 3168, AUSTRALIA and DAVID H. SOLOMON Department of Chemical and Biomolecular Engineering. University of Melbourne, Victoria 3010, AUSTRALIA Dr Graeme Moad CSIRO Molecular and Health Technologies Bayview Ave, Clayton, Victoria 3168 AUSTRALIA Email: [email protected] Prof David H. Solomon Department of Chemical and Biomolecular Engineering. University of Melbourne Victoria 3010 AUSTRALIA Email: [email protected] Contents Contents............................................................................................................................ v Index to Tables.............................................................................................................xvi Index to Figures ............................................................................................................ xx Preface to the First Edition .....................................................................................xxiii Preface to the Second Edition..................................................................................xxv Acknowledgments......................................................................................................xxvi 1 INTRODUCTION.................................................................................................. 1 1.1 References ........................................................................................................... -

A Comparative Study of the Formation of Aromatics In
A COMPARATIVE STUDY OF THE FORMATION OF AROMATICS IN RICH METHANE FLAMES DOPED BY UNSATURATED COMPOUNDS H.A. GUENICHE, J. BIET, P.A. GLAUDE, R. FOURNET, F. BATTIN-LECLERC* Département de Chimie-Physique des Réactions, Nancy Université, CNRS, 1 rue Grandville, BP 20451, 54001 NANCY Cedex, France * E-mail : [email protected] ; Tel.: 33 3 83 17 51 25 , Fax : 33 3 83 37 81 20 ABSTRACT For a better modeling of the importance of the different channels leading to the first aromatic ring, we have compared the structures of laminar rich premixed methane flames doped with several unsaturated hydrocarbons: allene and propyne, because they are precursors of propargyl radicals which are well known as having an important role in forming benzene, 1,3-butadiene to put in evidence a possible production of benzene due to reactions of C4 compounds, and, finally, cyclopentene which is a source of cyclopentadienylmethylene radicals which in turn are expected to easily isomerizes to give benzene. These flames have been stabilized on a burner at a pressure of 6.7 kPa (50 Torr) using argon as dilutant, for equivalence ratios (φ) from 1.55 to 1.79. A unique mechanism, including the formation and decomposition of benzene and toluene, has been used to model the oxidation of allene, propyne, 1,3-butadiene and cyclopentene. The main reaction pathways of aromatics formation have been derived from reaction rate and sensitivity analyses and have been compared for the three types of additives. These combined analyses and comparisons can only been performed when a unique mechanism is available for all the studied additives. -

Chapter 8 - Alkynes: an Introduction to Organic Synthesis
Chapter 8 - Alkynes: An Introduction to Organic Synthesis Draw structures corresponding to each of the following names. 1. ethynylcyclopropane Answer: CCH 2. 3,10-dimethyl-6-sec-butylcyclodecyne Answer: 3. 4-bromo-3,3-dimethyl-1-hexen-5-yne CH3 Br Answer: H 2C CH C CH C C H CH3 4. acetylene Answer: H CCH Provide names for each compound below. CH3 5. CH3C CCHCH2CH2CH3 Answer: 4-methyl-2-heptyne CH 3 6. CCH Answer: 1-ethynyl-2-methylcyclopentane Test Items for McMurry’s Organic Chemistry, Seventh Edition 59 The compound below has been isolated from the safflower plant. Consider its structure to answer the following questions. H H CCCCCCCC H H3C C C C H H C H H 7. What is the molecular formula for this natural product? Answer: C13H10 8. What is the degree of unsaturation for this compound? Answer: We can arrive at the degree of unsaturation for a structure in two ways. Since we know that the degree of unsaturation is the number of rings and/or multiple bonds in a compound, we can simply count them. There are three double bonds (3 degrees) and three triple bonds (six degrees), so the degree of unsaturation is 9. We can verify this by using the molecular formula, C13H10, to calculate a degree of unsaturation. The saturated 13-carbon compound should have the base formula C13H28, so (28 - 10) ÷ 2 = 18 ÷ 2 = 9. 9. Assign E or Z configuration to each of the double bonds in the compound. Answer: H H E CCCCCCCCE H H3C C C C H H C H H 10.