Identification of a Simplest Hypervalent Hydrogen Fluoride
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Courses of Study for Generic Elective 'B
COURSES OF STUDY FOR GENERIC ELECTIVE 'B. Sc, HODs' PROGRAMME IN "CHEMISTRY" GENERIC ELECTIVE All Four Generic Papers (One paper to be studied in each semester) of Chemistry to be studied by the Students of Other than Chemistry Honours. SEM- I Generic Elective Papers (GE-l) (Minor-Chemistry) (any four) for other Departments/ Disciplines: (Credit: 06 each) GE: ATOMIC STRUCTURE, BONDING, GENERAL ORGANIC CHEMISTRY & ALIPHATIC HYDROCARBONS (Credits: Theory-04, Practicals-02) Theory: 60 Lectures Section A: Inorganic Chemistry-I (30 Periods) Atomic Structure: Review of Bohr's theory and its limitations, dual behaviour of matter and radiation, de Broglie's relation, Heisenberg Uncertainty principle. Hydrogen atom spectra. Need of a new approach to Atomic structure. What is Quantum mechanics? Time independent Schrodinger equation and meaning of various terms in it. Significance of !fl and !fl2, Schrodinger equation for hydrogen atom. Radial and angular parts of the hydogenic wavefunctions (atomic orbitals) and their variations for Is, 2s, 2p, 3s, 3p and 3d orbitals (Only graphical representation). Radial and angular nodes and their significance. Radial distribution functions and the concept of the most probable distance with special reference to Is and 2s atomic orbitals. Significance of quantum numbers, orbital angular momentum and quantum numbers m I and m«. Shapes of s, p and d atomic orbitals, nodal planes. Discovery of spin, spin quantum number (s) and magnetic spin quantum number (ms). Rules for filling electrons in various orbitals, Electronic configurations of the atoms. Stability of half-filled and completely filled orbitals, concept of exchange energy. Relative energies of atomic orbitals, Anomalous electronic configurations. -

Topological Analysis of the Metal-Metal Bond: a Tutorial Review Christine Lepetit, Pierre Fau, Katia Fajerwerg, Myrtil L
Topological analysis of the metal-metal bond: A tutorial review Christine Lepetit, Pierre Fau, Katia Fajerwerg, Myrtil L. Kahn, Bernard Silvi To cite this version: Christine Lepetit, Pierre Fau, Katia Fajerwerg, Myrtil L. Kahn, Bernard Silvi. Topological analysis of the metal-metal bond: A tutorial review. Coordination Chemistry Reviews, Elsevier, 2017, 345, pp.150-181. 10.1016/j.ccr.2017.04.009. hal-01540328 HAL Id: hal-01540328 https://hal.sorbonne-universite.fr/hal-01540328 Submitted on 16 Jun 2017 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Topological analysis of the metal-metal bond: a tutorial review Christine Lepetita,b, Pierre Faua,b, Katia Fajerwerga,b, MyrtilL. Kahn a,b, Bernard Silvic,∗ aCNRS, LCC (Laboratoire de Chimie de Coordination), 205, route de Narbonne, BP 44099, F-31077 Toulouse Cedex 4, France. bUniversité de Toulouse, UPS, INPT, F-31077 Toulouse Cedex 4, i France cSorbonne Universités, UPMC, Univ Paris 06, UMR 7616, Laboratoire de Chimie Théorique, case courrier 137, 4 place Jussieu, F-75005 Paris, France Abstract This contribution explains how the topological methods of analysis of the electron density and related functions such as the electron localization function (ELF) and the electron localizability indicator (ELI-D) enable the theoretical characterization of various metal-metal (M-M) bonds (multiple M-M bonds, dative M-M bonds). -
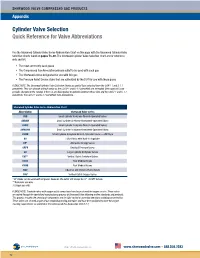
Cylinder Valve Selection Quick Reference for Valve Abbreviations
SHERWOOD VALVE COMPRESSED GAS PRODUCTS Appendix Cylinder Valve Selection Quick Reference for Valve Abbreviations Use the Sherwood Cylinder Valve Series Abbreviation Chart on this page with the Sherwood Cylinder Valve Selection Charts found on pages 73–80. The Sherwood Cylinder Valve Selection Chart are for reference only and list: • The most commonly used gases • The Compressed Gas Association primary outlet to be used with each gas • The Sherwood valves designated for use with this gas • The Pressure Relief Device styles that are authorized by the DOT for use with these gases PLEASE NOTE: The Sherwood Cylinder Valve Selection Charts are partial lists extracted from the CGA V-1 and S-1.1 pamphlets. They can change without notice as the CGA V-1 and S-1.1 pamphlets are amended. Sherwood will issue periodic changes to the catalog. If there is any discrepancy or question between these lists and the CGA V-1 and S-1.1 pamphlets, the CGA V-1 and S-1.1 pamphlets take precedence. Sherwood Cylinder Valve Series Abbreviation Chart Abbreviation Sherwood Valve Series AVB Small Cylinder Acetylene Wrench-Operated Valves AVBHW Small Cylinder Acetylene Handwheel-Operated Valves AVMC Small Cylinder Acetylene Wrench-Operated Valves AVMCHW Small Cylinder Acetylene Handwheel-Operated Valves AVWB Small Cylinder Acetylene Wrench-Operated Valves — WB Style BV Hi/Lo Valves with Built-in Regulator DF* Alternative Energy Valves GRPV Residual Pressure Valves GV Large Cylinder Acetylene Valves GVT** Vertical Outlet Acetylene Valves KVAB Post Medical Valves KVMB Post Medical Valves NGV Industrial and Chrome-Plated Valves YVB† Vertical Outlet Oxygen Valves 1 * DF Valves can be used with all gases; however, the outlet will always be ⁄4"–18 NPT female. -

Chapter 4, Lesson 5: Energy Levels, Electrons, and Ionic Bonding
Chapter 4, Lesson 5: Energy Levels, Electrons, and Ionic Bonding Key Concepts • The attractions between the protons and electrons of atoms can cause an electron to move completely from one atom to the other. • When an atom loses or gains an electron, it is called an ion. • The atom that loses an electron becomes a positive ion. • The atom that gains an electron becomes a negative ion. • A positive and negative ion attract each other and form an ionic bond. Summary Students will look at animations and make drawings of the ionic bonding of sodium chloride (NaCl). Students will see that both ionic and covalent bonding start with the attractions of pro- tons and electrons between different atoms. But in ionic bonding, electrons are transferred from one atom to the other and not shared like in covalent bonding. Students will use Styrofoam balls to make models of the ionic bonding in sodium chloride (salt). Objective Students will be able to explain the process of the formation of ions and ionic bonds. Evaluation The activity sheet will serve as the “Evaluate” component of each 5-E lesson plan. The activity sheets are formative assessments of student progress and understanding. A more formal summa- tive assessment is included at the end of each chapter. Safety Be sure you and the students wear properly fitting goggles. Materials for Each Group • Black paper • Salt • Cup with salt from evaporated saltwater • Magnifier • Permanent marker Materials for Each Student • 2 small Styrofoam balls • 2 large Styrofoam balls • 2 toothpicks ©2016 American Chemical Society Middle School Chemistry - www.middleschoolchemistry.com 337 Note: In an ionically bonded substance such as NaCl, the smallest ratio of positive and negative ions bonded together is called a “formula unit” rather than a “molecule.” Technically speaking, the term “molecule” refers to two or more atoms that are bonded together covalently, not ioni- cally. -

Chemical Forces Understanding the Relative Melting/Boiling Points of Two
Chapter 8 – Chemical Forces Understanding the relative melting/boiling points of two substances requires an understanding of the forces acting between molecules of those substances. These intermolecular forces are important for many additional reasons. For example, solubility and vapor pressure are governed by intermolecular forces. The same factors that give rise to intermolecular forces (e.g. bond polarity) can also have a profound impact on chemical reactivity. Chemical Forces Internuclear Distances and Atomic Radii There are four general methods of discussing interatomic distances: van der Waal’s, ionic, covalent, and metallic radii. We will discuss the first three in this section. Each has a unique perspective of the nature of the interaction between interacting atoms/ions. Van der Waal's Radii - half the distance between two nuclei of the same element in the solid state not chemically bonded together (e.g. solid noble gases). In general, the distance of separation between adjacent atoms (not bound together) in the solid state should be the sum of their van der Waal’s radii. F F van der Waal's radii F F Ionic Radii – Ionic radii were discussed in Chapter 4 and you should go back and review that now. One further thing is worth mentioning here. Evidence that bonding really exists and is attractive can be seen in ionic radii. For all simple ionic compounds, the ions attain noble gas configurations (e.g. in NaCl the Na+ ion is isoelectronic to neon and the Cl- ion is isoelectronic to argon). For the sodium chloride example just given, van der Waal’s radii would predict (Table 8.1, p. -

A Comparative Study of the Formation of Aromatics In
A COMPARATIVE STUDY OF THE FORMATION OF AROMATICS IN RICH METHANE FLAMES DOPED BY UNSATURATED COMPOUNDS H.A. GUENICHE, J. BIET, P.A. GLAUDE, R. FOURNET, F. BATTIN-LECLERC* Département de Chimie-Physique des Réactions, Nancy Université, CNRS, 1 rue Grandville, BP 20451, 54001 NANCY Cedex, France * E-mail : [email protected] ; Tel.: 33 3 83 17 51 25 , Fax : 33 3 83 37 81 20 ABSTRACT For a better modeling of the importance of the different channels leading to the first aromatic ring, we have compared the structures of laminar rich premixed methane flames doped with several unsaturated hydrocarbons: allene and propyne, because they are precursors of propargyl radicals which are well known as having an important role in forming benzene, 1,3-butadiene to put in evidence a possible production of benzene due to reactions of C4 compounds, and, finally, cyclopentene which is a source of cyclopentadienylmethylene radicals which in turn are expected to easily isomerizes to give benzene. These flames have been stabilized on a burner at a pressure of 6.7 kPa (50 Torr) using argon as dilutant, for equivalence ratios (φ) from 1.55 to 1.79. A unique mechanism, including the formation and decomposition of benzene and toluene, has been used to model the oxidation of allene, propyne, 1,3-butadiene and cyclopentene. The main reaction pathways of aromatics formation have been derived from reaction rate and sensitivity analyses and have been compared for the three types of additives. These combined analyses and comparisons can only been performed when a unique mechanism is available for all the studied additives. -

High Purity/Recovery Separation of Propylene from Propyne Using Anion Pillared Metal-Organic Framework: Application of Vacuum Swing Adsorption (VSA)
energies Article High Purity/Recovery Separation of Propylene from Propyne Using Anion Pillared Metal-Organic Framework: Application of Vacuum Swing Adsorption (VSA) Majeda Khraisheh 1, Fares AlMomani 1,* and Gavin Walker 2 1 Department of Chemical Engineering, College of Engineering, Qatar University, Doha P.O. Box 2713, Qatar; [email protected] 2 Department of Chemical Sciences, SSPC, Bernal Institute, University of Limerick, V94 T9PX Limerick, Ireland; [email protected] * Correspondence: [email protected]; Tel.: +974-4403-4140 Abstract: Propylene is one of the world’s most important basic olefin raw material used in the produc- tion of a vast array of polymers and other chemicals. The need for high purity grade of propylene is essential and traditionally achieved by the very energy-intensive cryogenic separation. In this study, 2− a pillared inorganic anion SIF6 was used as a highly selective C3H4 due to the square grid pyrazine- based structure. Single gas adsorption revealed a very high C3H4 uptake value (3.32, 3.12, 2.97 and 2.43 mmol·g−1 at 300, 320, 340 and 360 K, respectively). The values for propylene for the same tempera- tures were 2.73, 2.64, 2.31 and 1.84 mmol·g−1, respectively. Experimental results were obtained for the two gases fitted using Langmuir and Toth models. The former had a varied degree of representation of the system with a better presentation of the adsorption of the propylene compared to the propyne system. The Toth model regression offered a better fit of the experimental data over the entire range of pressures. -

Starter for Ten 3
Learn Chemistry Starter for Ten 3. Bonding Developed by Dr Kristy Turner, RSC School Teacher Fellow 2011-2012 at the University of Manchester, and Dr Catherine Smith, RSC School Teacher Fellow 2011-2012 at the University of Leicester This resource was produced as part of the National HE STEM Programme www.rsc.org/learn-chemistry Registered Charity Number 207890 3. BONDING 3.1. The nature of chemical bonds 3.1.1. Covalent dot and cross 3.1.2. Ionic dot and cross 3.1.3. Which type of chemical bond 3.1.4. Bonding summary 3.2. Covalent bonding 3.2.1. Co-ordinate bonding 3.2.2. Electronegativity and polarity 3.2.3. Intermolecular forces 3.2.4. Shapes of molecules 3.3. Properties and bonding Bonding answers 3.1.1. Covalent dot and cross Draw dot and cross diagrams to illustrate the bonding in the following covalent compounds. If you wish you need only draw the outer shell electrons; (2 marks for each correct diagram) 1. Water, H2O 2. Carbon dioxide, CO2 3. Ethyne, C2H2 4. Phosphoryl chloride, POCl3 5. Sulfuric acid, H2SO4 Bonding 3.1.1. 3.1.2. Ionic dot and cross Draw dot and cross diagrams to illustrate the bonding in the following ionic compounds. (2 marks for each correct diagram) 1. Lithium fluoride, LiF 2. Magnesium chloride, MgCl2 3. Magnesium oxide, MgO 4. Lithium hydroxide, LiOH 5. Sodium cyanide, NaCN Bonding 3.1.2. 3.1.3. Which type of chemical bond There are three types of strong chemical bonds; ionic, covalent and metallic. -

(12) United States Patent (10) Patent No.: US 8,198.491 B2 Masatoshi Et Al
USOO8198491 B2 (12) United States Patent (10) Patent No.: US 8,198.491 B2 Masatoshi et al. (45) Date of Patent: Jun. 12, 2012 (54) PROCESS FOR PREPARING 5,986,151 A 1 1/1999 Van Der Puy 2,3,3,3-TETRAFLUOROPROPENE AND $425, 23.99 Esthet al. 1,3,3,3-TETRAFLUOROPROPENE 7,833.434 B2 11/2010 Rao et al. 2005/0245773 Al 1 1/2005 Mukhopadhyay et al. (75) Inventors: Nose Masatoshi, Settsu (JP): Komatsu 2006/0258891 A1 1 1/2006 Mukhopadhyay et al. Yuzo, Settsu (JP); Sugiyama Akinari, 3.s! .k A. 239. SgO et stal al. Situ (JP); Shibanuma Takashi, Settsu 2009,0264689 A1 10, 2009 Rao et al. (JP) 2010/0200798 A1 8, 2010 Rao et al. (73) Assignee: Daikin Industries, Ltd., Osaka (JP) FOREIGN PATENT DOCUMENTS EP O 974 571 1, 2000 (*) Notice: Subject to any disclaimer, the term of this JP 63-211245 9, 1988 patent is extended or adjusted under 35 JP 11-14.0002 5, 1999 U.S.C. 154(b) by 0 days. JP 2007-320896 12/2007 WO 2008/OO2499 1, 2008 WO 2008/OO2500 1, 2008 (21) Appl. No.: 13/057,525 WO 2008/030440 3, 2008 (22) PCT Filed: Jul. 21, 2009 OTHER PUBLICATIONS International Search Report issued Mar. 3, 2010 in International (86). PCT No.: PCT/UP2O09/063314 (PCT) Application No. PCT/JP2009/063314. S371 (c)(1) PCT Written Opinion of the International Searching Authority issued (2), (4) Date:s Feb. 4, 2011 Mar.sia. 3, 2010 in International ((PCT) ) Application NoNo. PCT/JP2009/ Haszeldine et al., “Addition of FreeRadicals to Unsaturated Systems. -

Hydrogen Sulfide Capture: from Absorption in Polar Liquids to Oxide
Review pubs.acs.org/CR Hydrogen Sulfide Capture: From Absorption in Polar Liquids to Oxide, Zeolite, and Metal−Organic Framework Adsorbents and Membranes Mansi S. Shah,† Michael Tsapatsis,† and J. Ilja Siepmann*,†,‡ † Department of Chemical Engineering and Materials Science, University of Minnesota, 421 Washington Avenue SE, Minneapolis, Minnesota 55455-0132, United States ‡ Department of Chemistry and Chemical Theory Center, University of Minnesota, 207 Pleasant Street SE, Minneapolis, Minnesota 55455-0431, United States ABSTRACT: Hydrogen sulfide removal is a long-standing economic and environ- mental challenge faced by the oil and gas industries. H2S separation processes using reactive and non-reactive absorption and adsorption, membranes, and cryogenic distillation are reviewed. A detailed discussion is presented on new developments in adsorbents, such as ionic liquids, metal oxides, metals, metal−organic frameworks, zeolites, carbon-based materials, and composite materials; and membrane technologies for H2S removal. This Review attempts to exhaustively compile the existing literature on sour gas sweetening and to identify promising areas for future developments in the field. CONTENTS 4.1. Polymeric Membranes 9785 4.2. Membranes for Gas−Liquid Contact 9786 1. Introduction 9755 4.3. Ceramic Membranes 9789 2. Absorption 9758 4.4. Carbon-Based Membranes 9789 2.1. Alkanolamines 9758 4.5. Composite Membranes 9790 2.2. Methanol 9758 N 5. Cryogenic Distillation 9790 2.3. -Methyl-2-pyrrolidone 9758 6. Outlook and Perspectives 9791 2.4. Poly(ethylene glycol) Dimethyl Ether 9759 Author Information 9792 2.5. Sulfolane and Diisopropanolamine 9759 Corresponding Author 9792 2.6. Ionic Liquids 9759 ORCID 9792 3. Adsorption 9762 Notes 9792 3.1. -

The Different Types of Bonds Atoms Form Bonds with Other Atoms in Order to Have a Full Outer Shell of Electrons Like the Noble Gases
Reading- The Different Types of Bonds Atoms form bonds with other atoms in order to have a full outer shell of electrons like the noble gases. If an atom has too few or too many valence electrons it will have to gain, lose, or share those outer electrons with another atom in order to become “happy” or in chemistry terms, more stable. There are many types of chemical bonds that can form, however the 3 main types are: ionic, covalent, and metallic bonds. You must become familiar with how they work and the differences between the 3 types. I. Ionic bonding: Model 1 is a description of what chemists call ionic bonding. Ionic bonding occurs strictly between metal and nonmetal atoms. In ionic bonding some of the valence electrons of a metal atom are transferred to a nonmetal atom so that each atom ends up with a noble gas configuration. Usually one, two, or three electrons are transferred from one atom to another. This transfer of an electron causes the metal atom to have a net positive charge (+) and the nonmetal atom to have a net negative charge (-). The individual atoms in ionic solids are referred to as ions because of their charges. These opposite charges are attracted to one another. On the right is a drawing of a chunk of salt, NaCl, a very common ionic substance. Notice how the sodium and chloride ions alternate throughout the structure. The positive and negative ions alternating in three dimensions make the solid quite strong because of their strong attractions to one another. -

Rich Methane Premixed Laminar Flames Doped with Light Unsaturated
Combustion and Flame 146 (2006) 620–634 www.elsevier.com/locate/combustflame Rich methane premixed laminar flames doped with light unsaturated hydrocarbons I. Allene and propyne H.A. Gueniche, P.A. Glaude, G. Dayma, R. Fournet, F. Battin-Leclerc ∗ Département de Chimie-Physique des Réactions, Nancy University, CNRS, ENSIC, 1 rue Grandville, BP 20451, 54001 Nancy Cedex, France Received 9 February 2006; received in revised form 16 June 2006; accepted 3 July 2006 Available online 4 August 2006 Abstract The structure of three laminar premixed rich flames has been investigated: a pure methane flame and two methane flames doped by allene and propyne, respectively. The gases of the three flames contain 20.9% (mo- lar) of methane and 33.4% of oxygen, corresponding to an equivalence ratio of 1.25 for the pure methane flame. In both doped flames, 2.49% of C3H4 was added, corresponding to a ratio C3H4/CH4 of 12% and an equivalence ra- tio of 1.55. The three flames have been stabilized on a burner at a pressure of 6.7 kPa using argon as dilutant, with a gas velocity at the burner of 36 cm/s at 333 K. The concentration profiles of stable species were measured by gas chromatography after sampling with a quartz microprobe. Quantified species included carbon monoxide and diox- ide, methane, oxygen, hydrogen, ethane, ethylene, acetylene, propyne, allene, propene, propane, 1,2-butadiene, 1,3-butadiene, 1-butene, isobutene, 1-butyne, vinylacetylene, and benzene. The temperature was measured using a PtRh (6%)–PtRh (30%) thermocouple settled inside the enclosure and ranged from 700 K close to the burner up to 1850 K.