Biochemical and Physiological Investigations on Adenosine 5’
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

United States Patent 19 11 Patent Number: 5,780,253 Subramanian Et Al
III USOO5780253A United States Patent 19 11 Patent Number: 5,780,253 Subramanian et al. (45) Date of Patent: Jul. 14, 1998 54 SCREENING METHOD FOR DETECTION OF 4.433.999 2/1984 Hyzak ....................................... 71.03 HERBCDES 4.6–552 2/1987 Anoti et al. if O3. 4,802,912 2/1989 Baker ........................................ 7/103 Inventors: Wenkiteswaran Subramanian Danville: Anne G. Toschi. Burlingame. OTHERTHER PPUBLICATION CATIONS both of Calif. Heim et al. Pesticide Biochem & Physiol; vol. 53, pp. 138-145 (1995). 73) Assignee: Sandoz Ltd., Basel. Switzerland Hatch. MD.: Phytochem. vol. 6... pp. 115 to 119, (1967). Haworth et al. J. Agric. Food Chem, vol. 38, pp. 1271-1273. 21 Appl. No.:752.990 1990. Nishimura et al: Phytochem: vol. 34, pp. 613-615. (1993). 22 Filed: Nov. 21, 1996 Primary Examiner-Louise N. Leary Related U.S. Application Data Attorney, Agent, or Firm-Lynn Marcus-Wyner: Michael P. Morris 63 Continuation of Ser. No. 434.826, May 4, 1995, abandoned. 6 57 ABSTRACT 51 Int. Cl. ............................... C12Q 1/48: C12Q 1/32: C12Q 1/37; C12O 1/00 This invention relates to novel screening methods for iden 52 U.S. Cl. ................................. 435/15:435/18: 435/26: tifying compounds that specifically inhibit a biosynthetic 435/23: 435/4, 536/23.6:536/23.2:536/24.3 pathway in plants. Enzymes which are specifically affected 536/26.11:536/26.12:536/26.13 by the novel screening method include plant purine biosyn 58 Field of Search .................................. 435/15, 8, 26, thetic pathway enzymes and particularly the enzymes 435/23 4: 536/23.6, 23.2, 24.3, 26.1, involved in the conversion of inosine monophosphate to 26.12, 26.13 adenosine monophosphate and inosine monophosphate to guanosine monophosphate. -

35 Disorders of Purine and Pyrimidine Metabolism
35 Disorders of Purine and Pyrimidine Metabolism Georges van den Berghe, M.- Françoise Vincent, Sandrine Marie 35.1 Inborn Errors of Purine Metabolism – 435 35.1.1 Phosphoribosyl Pyrophosphate Synthetase Superactivity – 435 35.1.2 Adenylosuccinase Deficiency – 436 35.1.3 AICA-Ribosiduria – 437 35.1.4 Muscle AMP Deaminase Deficiency – 437 35.1.5 Adenosine Deaminase Deficiency – 438 35.1.6 Adenosine Deaminase Superactivity – 439 35.1.7 Purine Nucleoside Phosphorylase Deficiency – 440 35.1.8 Xanthine Oxidase Deficiency – 440 35.1.9 Hypoxanthine-Guanine Phosphoribosyltransferase Deficiency – 441 35.1.10 Adenine Phosphoribosyltransferase Deficiency – 442 35.1.11 Deoxyguanosine Kinase Deficiency – 442 35.2 Inborn Errors of Pyrimidine Metabolism – 445 35.2.1 UMP Synthase Deficiency (Hereditary Orotic Aciduria) – 445 35.2.2 Dihydropyrimidine Dehydrogenase Deficiency – 445 35.2.3 Dihydropyrimidinase Deficiency – 446 35.2.4 Ureidopropionase Deficiency – 446 35.2.5 Pyrimidine 5’-Nucleotidase Deficiency – 446 35.2.6 Cytosolic 5’-Nucleotidase Superactivity – 447 35.2.7 Thymidine Phosphorylase Deficiency – 447 35.2.8 Thymidine Kinase Deficiency – 447 References – 447 434 Chapter 35 · Disorders of Purine and Pyrimidine Metabolism Purine Metabolism Purine nucleotides are essential cellular constituents 4 The catabolic pathway starts from GMP, IMP and which intervene in energy transfer, metabolic regula- AMP, and produces uric acid, a poorly soluble tion, and synthesis of DNA and RNA. Purine metabo- compound, which tends to crystallize once its lism can be divided into three pathways: plasma concentration surpasses 6.5–7 mg/dl (0.38– 4 The biosynthetic pathway, often termed de novo, 0.47 mmol/l). starts with the formation of phosphoribosyl pyro- 4 The salvage pathway utilizes the purine bases, gua- phosphate (PRPP) and leads to the synthesis of nine, hypoxanthine and adenine, which are pro- inosine monophosphate (IMP). -

SUPPY Liglucosexlmtdh
US 20100314248A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2010/0314248 A1 Worden et al. (43) Pub. Date: Dec. 16, 2010 (54) RENEWABLE BOELECTRONIC INTERFACE Publication Classification FOR ELECTROBOCATALYTC REACTOR (51) Int. Cl. (76) Inventors: Robert M. Worden, Holt, MI (US); C25B II/06 (2006.01) Brian L. Hassler, Lake Orion, MI C25B II/2 (2006.01) (US); Lawrence T. Drzal, Okemos, GOIN 27/327 (2006.01) MI (US); Ilsoon Lee, Okemo s, MI BSD L/04 (2006.01) (US) C25B 9/00 (2006.01) (52) U.S. Cl. ............... 204/403.14; 204/290.11; 204/400; Correspondence Address: 204/290.07; 427/458; 204/252: 977/734; PRICE HENEVELD COOPER DEWITT & LIT 977/742 TON, LLP 695 KENMOOR, S.E., PO BOX 2567 (57) ABSTRACT GRAND RAPIDS, MI 495.01 (US) An inexpensive, easily renewable bioelectronic device useful for bioreactors, biosensors, and biofuel cells includes an elec (21) Appl. No.: 12/766,169 trically conductive carbon electrode and a bioelectronic inter face bonded to a surface of the electrically conductive carbon (22) Filed: Apr. 23, 2010 electrode, wherein the bioelectronic interface includes cata lytically active material that is electrostatically bound directly Related U.S. Application Data or indirectly to the electrically conductive carbon electrode to (60) Provisional application No. 61/172,337, filed on Apr. facilitate easy removal upon a change in pH, thereby allowing 24, 2009. easy regeneration of the bioelectronic interface. 7\ POWER 1 - SUPPY|- LIGLUCOSEXLMtDH?till pi 6.0 - esses&aaaas-exx-xx-xx-xx-xxxxixax-e- Patent Application Publication Dec. 16, 2010 Sheet 1 of 18 US 2010/0314248 A1 Potential (nV) Patent Application Publication Dec. -

Nucleotide Metabolism Pathway: the Achilles' Heel for Bacterial Pathogens
REVIEW ARTICLES Nucleotide metabolism pathway: the achilles’ heel for bacterial pathogens Sujata Kumari1,2,* and Prajna Tripathi1,3 1National Institute of Immunology, New Delhi 110 067, India 2Present address: Department of Zoology, Magadh Mahila College, Patna University, Patna 800 001, India 3Present address: Institute of Molecular Medicine, Jamia Hamdard, New Delhi 110 062, India de novo pathway, the nucleotides are synthesized from Pathogens exploit their host to extract nutrients for their survival. They occupy a diverse range of host simple precursor molecules. In the salvage pathway, the niches during infection which offer variable nutrients preformed nucleobases or nucleosides which are present accessibility. To cause a successful infection a patho- in the cell or transported from external environmental gen must be able to acquire these nutrients from the milieu to the cell are utilized to form nucleotides. host as well as be able to synthesize the nutrients on its own, if required. Nucleotides are the essential me- tabolite for a pathogen and also affect the pathophysi- Purine biosynthesis pathway ology of infection. This article focuses on the role of nucleotide metabolism of pathogens during infection The purine biosynthesis pathway is universally conserved in a host. Nucleotide metabolism and disease pathoge- in living organisms (Figure 1). As an example, we here nesis are closely related in various pathogens. Nucleo- present the pathway derived from well-studied Gram- tides, purines and pyrimidines, are biosynthesized by positive bacteria Lactococcus lactis. In the de novo the de novo and salvage pathways. Whether the patho- pathway the purine nucleotides are synthesized from sim- gen will employ the de novo or salvage pathway dur- ple molecules such as phosphoribosyl pyrophosphate ing infection is dependent on various factors, like (PRPP), amino acids, CO2 and NH3 by a series of enzy- availability of nucleotides, energy condition and pres- matic reactions. -

Annual Symposium of the Society for the Study of Inborn Errors of Metabolism Birmingham, UK, 4 – 7 September 2012
J Inherit Metab Dis (2012) 35 (Suppl 1):S1–S182 DOI 10.1007/s10545-012-9512-z ABSTRACTS Annual Symposium of the Society for the Study of Inborn Errors of Metabolism Birmingham, UK, 4 – 7 September 2012 This supplement was not sponsored by outside commercial interests. It was funded entirely by the SSIEM. 01. Amino Acids and PKU O-002 NATURAL INHIBITORS OF CARNOSINASE (CN1) O-001 Peters V1 ,AdelmannK2 ,YardB2 , Klingbeil K1 ,SchmittCP1 , Zschocke J3 3-HYDROXYISOBUTYRIC ACID DEHYDROGENASE DEFICIENCY: 1Zentrum für Kinder- und Jugendmedizin de, Heidelberg, Germany IDENTIFICATION OF A NEW INBORN ERROR OF VALINE 2Universitätsklinik, Mannheim, Germany METABOLISM 3Humangenetik, Innsbruck, Austria Wanders RJA1 , Ruiter JPN1 , Loupatty F.1 , Ferdinandusse S.1 , Waterham HR1 , Pasquini E.1 Background: Carnosinase degrades histidine-containing dipeptides 1Div Metab Dis, Univ Child Hosp, Amsterdam, Netherlands such as carnosine and anserine which are known to have several protective functions, especially as antioxidant agents. We recently Background, Objectives: Until now only few patients with an established showed that low carnosinase activities protect from diabetic nephrop- defect in the valine degradation pathway have been identified. Known athy, probably due to higher histidine-dipeptide concentrations. We deficiencies include 3-hydroxyisobutyryl-CoA hydrolase deficiency and now characterized the carnosinase metabolism in children and identi- methylmalonic semialdehyde dehydrogenase (MMSADH) deficiency. On fied natural inhibitors of carnosinase. the other hand many patients with 3-hydroxyisobutyric aciduria have been Results: Whereas serum carnosinase activity and protein concentrations described with a presumed defect in the valine degradation pathway. To correlate in adults, children have lower carnosinase activity although pro- identify the enzymatic and molecular defect in a group of patients with 3- tein concentrations were within the same level as for adults. -

Developmental Disorder Associated with Increased Cellular Nucleotidase Activity (Purine-Pyrimidine Metabolism͞uridine͞brain Diseases)
Proc. Natl. Acad. Sci. USA Vol. 94, pp. 11601–11606, October 1997 Medical Sciences Developmental disorder associated with increased cellular nucleotidase activity (purine-pyrimidine metabolismyuridineybrain diseases) THEODORE PAGE*†,ALICE YU‡,JOHN FONTANESI‡, AND WILLIAM L. NYHAN‡ Departments of *Neurosciences and ‡Pediatrics, University of California at San Diego, La Jolla, CA 92093 Communicated by J. Edwin Seegmiller, University of California at San Diego, La Jolla, CA, August 7, 1997 (received for review June 26, 1997) ABSTRACT Four unrelated patients are described with a represent defects of purine metabolism, although no specific syndrome that included developmental delay, seizures, ataxia, enzyme abnormality has been identified in these cases (6). In recurrent infections, severe language deficit, and an unusual none of these disorders has it been possible to delineate the behavioral phenotype characterized by hyperactivity, short mechanism through which the enzyme deficiency produces the attention span, and poor social interaction. These manifesta- neurological or behavioral abnormalities. Therapeutic strate- tions appeared within the first few years of life. Each patient gies designed to treat the behavioral and neurological abnor- displayed abnormalities on EEG. No unusual metabolites were malities of these disorders by replacing the supposed deficient found in plasma or urine, and metabolic testing was normal metabolites have not been successful in any case. except for persistent hypouricosuria. Investigation of purine This report describes four unrelated patients in whom and pyrimidine metabolism in cultured fibroblasts derived developmental delay, seizures, ataxia, recurrent infections, from these patients showed normal incorporation of purine speech deficit, and an unusual behavioral phenotype were bases into nucleotides but decreased incorporation of uridine. -

Supplements Inference of Cancer Mechanisms Through Computational Systems Analysis
Electronic Supplementary Material (ESI) for Molecular BioSystems. This journal is © The Royal Society of Chemistry 2017 Supplements Inference of Cancer Mechanisms through Computational Systems Analysis Zhen Qi and Eberhard O. Voit Mathematical model of purine metabolism in human. A published kinetic model of human purine metabolism was used as a computational platform; it consists of 16 ordinary differential equations with 37 fluxes 1, 2. In the literature, the fluxes were formulated either as traditional Michaelis-Menten kinetics or as power law functions under the tenets of Biochemical Systems Theory 3. Here we chose the latter. Many parameters were obtained from experimental and clinical data in humans, and the remaining values were estimated using biological constraints, such as the ratio of adenine and guanine in nucleic acids, which is approximately 3/2, or the fact that normal subjects excrete about 420 mg per day of UA in urine. A detailed analysis of the steady-state properties of the mathematical model demonstrated that the steady state is stable and robust. Analysis also showed that the model is not sensitive to parameter changes. Simulations of normal and pathological perturbations of purine metabolism yielded consistent results with some representative biochemical and clinical observations. All these analyses are described in great detail in the literature 3. Implementation of enzyme activities altered by cancer. Weber discovered several changes in the enzyme activities of purine metabolism in human renal carcinoma cells 4. The affected enzymes and their fold changes compared to normal kidney cells are listed in Table S4. These alterations are expected to result in changed metabolite levels between normal human cells and human renal cell carcinoma, which were computed with the mathematical model, and the values at the corresponding steady state are shown in Table 1 in the Text. -

Open Thesis Final Draft Janowitz.Pdf
THE PENNSYLVANIA STATE UNIVERSITY SCHREYER HONORS COLLEGE DEPARTMENT OF BIOCHEMISTRY AND MOLECULAR BIOLOGY MODELING THE REPRODUCTIVE PHENOTYPES OF ADENYLOSUCCINATE LYASE DEFICIENCY IN C. ELEGANS HALEY JANOWITZ FALL 2017 A thesis submitted in partial fulfillment of the requirements for a baccalaureate degree in Biochemistry and Molecular Biology with honors in Biochemistry and Molecular Biology Reviewed and approved* by the following: Wendy Hanna-Rose Interim Department Head, Biochemistry and Molecular Biology Associate Professor of Biochemistry and Molecular Biology Thesis Supervisor Sarah Ades Associate Professor of Biochemistry and Molecular Biology Honors Adviser * Signatures are on file in the Schreyer Honors College. i ABSTRACT Mutations in enzymes that function in purine metabolism result in human syndromes with a wide variety of symptoms. Adenylosuccinate lyase deficiency (ASLD) is characterized by the decrease in function of adenylosuccinate lyase (ADSL), a bi-functional enzyme within de novo purine biosynthesis. In humans, this syndrome is characterized by neuronal, developmental, and metabolic defects which include symptoms of seizures, encephalopathy, psychomotor retardation, and autistic features. The molecular mechanisms driving this disease are currently unknown. I report on the reproductive phenotypes associated with a knockdown of ADSL in Caenorhabditis elegans. I identify sterility in animals with significant knockdown of adsl-1 expression, as well as embryonic lethality and oogenesis defects associated with a partial knockdown of adsl-1 expression. Using supplementation, I correlate these phenotypes with a decreased flux through de novo purine biosynthesis. Although reproductive phenotypes are not directly correlated with human symptoms, these findings still have impact on the role of purines in the development of patients with ASLD. -

Diagnose a Broad Range of Metabolic Disorders with a Single Test, Global
PEDIATRIC Assessing or diagnosing a metabolic disorder commonly requires several tests. Global Metabolomic Assisted Pathway Screen, commonly known as Global MAPS, is a unifying test GLOBAL MAPS™ for analyzing hundreds of metabolites to identify changes Global Metabolomic or irregularities in biochemical pathways. Let Global MAPS Assisted Pathway Screen guide you to an answer. Diagnose a broad range of metabolic disorders with a single test, Global MAPS Global MAPS is a large scale, semi-quantitative metabolomic profiling screen that analyzes disruptions in both individual analytes and pathways related to biochemical abnormalities. Using state-of-the-art technologies, Global Metabolomic Assisted Pathway Screen (Global MAPS) provides small molecule metabolic profiling to identify >700 metabolites in human plasma, urine, or cerebrospinal fluid. Global MAPS identifies inborn errors of metabolism (IEMs) that would ordinarily require many different tests. This test defines biochemical pathway errors not currently detected by routine clinical or genetic testing. IEMs are inherited metabolic disorders that prevent the body from converting one chemical compound to another or from transporting a compound in or out of a cell. NORMAL PROCESS METABOLIC ERROR These processes are necessary for essentially all bodily functions. Most IEMs are caused by defects in the enzymes that help process nutrients, which result in an accumulation of toxic substances or a deficiency of substances needed for normal body function. Making a swift, accurate diagnosis -

O O2 Enzymes Available from Sigma Enzymes Available from Sigma
COO 2.7.1.15 Ribokinase OXIDOREDUCTASES CONH2 COO 2.7.1.16 Ribulokinase 1.1.1.1 Alcohol dehydrogenase BLOOD GROUP + O O + O O 1.1.1.3 Homoserine dehydrogenase HYALURONIC ACID DERMATAN ALGINATES O-ANTIGENS STARCH GLYCOGEN CH COO N COO 2.7.1.17 Xylulokinase P GLYCOPROTEINS SUBSTANCES 2 OH N + COO 1.1.1.8 Glycerol-3-phosphate dehydrogenase Ribose -O - P - O - P - O- Adenosine(P) Ribose - O - P - O - P - O -Adenosine NICOTINATE 2.7.1.19 Phosphoribulokinase GANGLIOSIDES PEPTIDO- CH OH CH OH N 1 + COO 1.1.1.9 D-Xylulose reductase 2 2 NH .2.1 2.7.1.24 Dephospho-CoA kinase O CHITIN CHONDROITIN PECTIN INULIN CELLULOSE O O NH O O O O Ribose- P 2.4 N N RP 1.1.1.10 l-Xylulose reductase MUCINS GLYCAN 6.3.5.1 2.7.7.18 2.7.1.25 Adenylylsulfate kinase CH2OH HO Indoleacetate Indoxyl + 1.1.1.14 l-Iditol dehydrogenase L O O O Desamino-NAD Nicotinate- Quinolinate- A 2.7.1.28 Triokinase O O 1.1.1.132 HO (Auxin) NAD(P) 6.3.1.5 2.4.2.19 1.1.1.19 Glucuronate reductase CHOH - 2.4.1.68 CH3 OH OH OH nucleotide 2.7.1.30 Glycerol kinase Y - COO nucleotide 2.7.1.31 Glycerate kinase 1.1.1.21 Aldehyde reductase AcNH CHOH COO 6.3.2.7-10 2.4.1.69 O 1.2.3.7 2.4.2.19 R OPPT OH OH + 1.1.1.22 UDPglucose dehydrogenase 2.4.99.7 HO O OPPU HO 2.7.1.32 Choline kinase S CH2OH 6.3.2.13 OH OPPU CH HO CH2CH(NH3)COO HO CH CH NH HO CH2CH2NHCOCH3 CH O CH CH NHCOCH COO 1.1.1.23 Histidinol dehydrogenase OPC 2.4.1.17 3 2.4.1.29 CH CHO 2 2 2 3 2 2 3 O 2.7.1.33 Pantothenate kinase CH3CH NHAC OH OH OH LACTOSE 2 COO 1.1.1.25 Shikimate dehydrogenase A HO HO OPPG CH OH 2.7.1.34 Pantetheine kinase UDP- TDP-Rhamnose 2 NH NH NH NH N M 2.7.1.36 Mevalonate kinase 1.1.1.27 Lactate dehydrogenase HO COO- GDP- 2.4.1.21 O NH NH 4.1.1.28 2.3.1.5 2.1.1.4 1.1.1.29 Glycerate dehydrogenase C UDP-N-Ac-Muramate Iduronate OH 2.4.1.1 2.4.1.11 HO 5-Hydroxy- 5-Hydroxytryptamine N-Acetyl-serotonin N-Acetyl-5-O-methyl-serotonin Quinolinate 2.7.1.39 Homoserine kinase Mannuronate CH3 etc. -
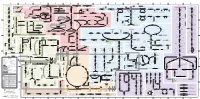
Fullsubwaymap221.Pdf
A B C D E F G H I J K L M Aldose reductase Sorbitol dehydrogenase Cholesterol synthesis Glycogen and galactose metabolism (branch point only) (debranching) glucose sorbitol fructose Aromatic amino acid metabolism Purine synthesis Pyrimidine synthesis glycogen (n+1) phenylacetate triiodothyronine (T3) dopaquinone melanin + + thyroxine (T4) (multiple steps) (many cycles) P 4' NADPH NADP NAD NADH acetyl-CoA x2 i Debranching enzyme 3' - α-1,4 bonds Aromatic amino acid 5-phosphoribosyl pyrophosphate (PRPP) HCO B 2' P 3 (branching) Glycogen 6 i (multiple steps) O H O (multiple steps) Tyrosinase O H O 1' 2 2 2 2 B 1 2 3 4 5 6 7 8 9 10 Pentose phosphate pathway Phenylalanine Tyrosine decarboxylase 6 Dopamine B Thiolase phosphorylase -5 -4 -3 -2 -1 0 1 2 3 4 Transaminase 6 glutamine 2 ATP (many cycles) Glycogen Glucose-6-phosphatase Dehydrogenase hydroxylase hydroxylase (DOPA decarboxylase) β-hydroxylase Methyltransferase 10 UDP 4:4 Transferase ATP glutathione (reduced) H O phenyllactate phenylpyruvate phenylalanine tyrosine dihydroxy- dopamine Glutamine PRPP CoA synthase Hexokinase or (in endoplasmic reticulum) 2 2 norepinephrine epinephrine glutamine Carbamoyl 9 1' phenylanine amidotransferase (GPAT) glutamate glycogen (n) Glutathione reductase Glutathione peroxidase + phosphate glycogen (n) -5 -4 -3 -2 -1 0 1 2 3 4 2' 3' 4' glucokinase 8 NADP NADPH glutamate α-ketoglutarate CO2 O H O Glycosyl (4:6) UDP-glucose ADP (L-DOPA) 2 2 synthetase II glutathione (oxidized) 2 H O α-ketoglutarate PPi glutamate acetoacetyl-CoA 7 1 Glucose -1,6-Glucosidase -

Multi-Omics Profiling of CHO Parental Hosts Reveals Cell Line
bioRxiv preprint doi: https://doi.org/10.1101/532150; this version posted January 31, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Multi-omics profiling of CHO parental hosts reveals 2 cell line-specific variations in bioprocessing traits 3 4 Meiyappan Lakshmanan1, Yee Jiun Kok1, Alison P. Lee1, Sarantos Kyriakopoulos1, Hsueh Lee 5 Lim1, Gavin Teo1, Swan Li Poh1, Wen Qin Tang1, Jongkwang Hong1, Andy Hee-Meng Tan1, 6 Xuezhi Bi1, Ying Swan Ho1, Peiqing Zhang1, Say Kong Ng1, Dong-Yup Lee1,2 7 8 1Bioprocessing Technology Institute, Agency for Science, Technology and Research (A*STAR), 9 20 Biopolis Way, #06-01, Centros 138668, Singapore 10 2School of Chemical Engineering, Sungkyunkwan University, 2066, Seobu-ro, Jangan-gu, 11 Suwon-si, Gyeonggi-do 16419, Republic of Korea; telephone: +82-31-290-7253; fax: +82-31- 12 290-7272; e-mail: [email protected] 13 14 15 16 17 18 19 Correspondence to: D.-Y. Lee 20 Contract grant sponsor: Agency for Science, Technology and Research, Singapore 21 Contract grant sponsor: Next-Generation BioGreen 21 Program, Rural Development 22 Administration, Republic of Korea 23 Contract grant number: PJ01334605 24 25 Running Head: Multi-omics characterization of CHO parental cell lines 1 bioRxiv preprint doi: https://doi.org/10.1101/532150; this version posted January 31, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity.