A High-Throughput Screening Platform for in Vitro Elastic Fiber
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Vocabulario De Morfoloxía, Anatomía E Citoloxía Veterinaria
Vocabulario de Morfoloxía, anatomía e citoloxía veterinaria (galego-español-inglés) Servizo de Normalización Lingüística Universidade de Santiago de Compostela COLECCIÓN VOCABULARIOS TEMÁTICOS N.º 4 SERVIZO DE NORMALIZACIÓN LINGÜÍSTICA Vocabulario de Morfoloxía, anatomía e citoloxía veterinaria (galego-español-inglés) 2008 UNIVERSIDADE DE SANTIAGO DE COMPOSTELA VOCABULARIO de morfoloxía, anatomía e citoloxía veterinaria : (galego-español- inglés) / coordinador Xusto A. Rodríguez Río, Servizo de Normalización Lingüística ; autores Matilde Lombardero Fernández ... [et al.]. – Santiago de Compostela : Universidade de Santiago de Compostela, Servizo de Publicacións e Intercambio Científico, 2008. – 369 p. ; 21 cm. – (Vocabularios temáticos ; 4). - D.L. C 2458-2008. – ISBN 978-84-9887-018-3 1.Medicina �������������������������������������������������������������������������veterinaria-Diccionarios�������������������������������������������������. 2.Galego (Lingua)-Glosarios, vocabularios, etc. políglotas. I.Lombardero Fernández, Matilde. II.Rodríguez Rio, Xusto A. coord. III. Universidade de Santiago de Compostela. Servizo de Normalización Lingüística, coord. IV.Universidade de Santiago de Compostela. Servizo de Publicacións e Intercambio Científico, ed. V.Serie. 591.4(038)=699=60=20 Coordinador Xusto A. Rodríguez Río (Área de Terminoloxía. Servizo de Normalización Lingüística. Universidade de Santiago de Compostela) Autoras/res Matilde Lombardero Fernández (doutora en Veterinaria e profesora do Departamento de Anatomía e Produción Animal. -

Flavio Akira Sakae Distribuição Das Fibras Colágenas E Do Sistema De
Flavio Akira Sakae Distribuição das fibras colágenas e do sistema de fibras elásticas na camada superficial da lâmina própria da prega vocal com edema de Reinke Tese apresentada à Faculdade de Medicina da Universidade de São Paulo para obtenção do título de Doutor em Ciências Área de concentração: Otorrinolaringologia Orientador: Prof. Dr. Domingos Hiroshi Tsuji São Paulo 2008 Dados Internacionais de Catalogação na Publicação (CIP) Preparada pela Biblioteca da Faculdade de Medicina da Universidade de São Paulo Óreprodução autorizada pelo autor Sakae, Flavio Akira Distribuição das fibras colágenas e do sistema de fibras elásticas na camada superficial da lâmina própria da prega vocal com edema de Reinke / Flavio Akira Sakae. -- São Paulo, 2008. Tese(doutorado)--Faculdade de Medicina da Universidade de São Paulo. Departamento de Oftalmologia e Otorrinolaringologia. Área de concentração: Otorrinolaringologia. Orientador: Domingos Hiroshi Tsuji. Descritores: 1.Edema laríngeo 2.Colágeno 3.Tecido elástico 4.Membrana mucosa 5.Cordas vocais USP/FM/SBD-146/08 "O único homem que está isento de erros, é aquele que não arrisca acertar." Albert Einstein Dedicatória Aos meus queridos pais, Masao e Junko, por tudo que fazem por mim, pelo apoio incondicional e amor eterno. São os meus ídolos. A minha esposa, Renata, amor da minha vida, pela alegria de viver, companheirismo e incentivo constante. A minha irmã, Cristiane, por ter contribuído em todos os passos de minha vida. Agradecimentos Ao meu orientador, Prof. Dr. Domingos Hiroshi Tsuji pela oportunidade e apoio na concretização deste sonho. Sua amizade e franqueza foram essenciais na elaboração deste trabalho. É o grande mestre. Ao Prof. Dr. -

Elastic Fiber Production in Cardiovascular Tissue-Equivalents
Matrix Biology 22 (2003) 339–350 Elastic fiber production in cardiovascular tissue-equivalents Jennifer L. Long, Robert T. Tranquillo* Department of Chemical Engineering & Materials Science and Department of Biomedical Engineering, 7-114 BSBE, 312 Church St SE, University of Minnesota, Minneapolis, MN 55455, USA Received 10 January 2003; received in revised form 30 April 2003; accepted 30 April 2003 Abstract Elastic fiber incorporation is critical to the success of tissue-engineered arteries and heart valves. Elastic fibers have not yet been observed in tissue-engineered replacements fabricated in vitro with smooth muscle cells. Here, rat smooth muscle cells (SMC) or human dermal fibroblasts (HDF) remodeled collagen or fibrin gels for 4 weeks as the basis for a completely biological cardiovascular tissue replacement. Immunolabeling, alkaline extraction and amino acid analysis identified and quantified elastin. Organized elastic fibers formed when neonatal SMC were cultured in fibrin gel. Fibrillin-1 deposition occurred but elastin was detected in regions without fibrillin-1, indicating that a microfibril template is not required for elastic fiber formation within fibrin. Collagen did not support substantial elastogenesis by SMC. The quantity of crosslinked elastic fibers was enhanced by treatment with TGF-b1 and insulin, concomitant with increased collagen production. These additives overcame ascorbate’s inhibition of elastogenesis in fibrin. The elasticfibers that formed in fibrin treated with TGF- b1 and insulin contained crosslinks, as evidenced by the presence of desmosine and an altered elastin labeling pattern when b-aminopropionitrile (BAPN) was added. These findings indicate that in vitro elastogenesis can be achieved in tissue engineering applications, and they suggest a physiologically relevant model system for the study of three-dimensional elastic structures. -
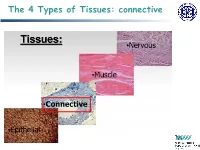
The 4 Types of Tissues: Connective
The 4 Types of Tissues: connective Connective Tissue General structure of CT cells are dispersed in a matrix matrix = a large amount of extracellular material produced by the CT cells and plays a major role in the functioning matrix component = ground substance often crisscrossed by protein fibers ground substance usually fluid, but it can also be mineralized and solid (bones) CTs = vast variety of forms, but typically 3 characteristic components: cells, large amounts of amorphous ground substance, and protein fibers. Connective Tissue GROUND SUBSTANCE In connective tissue, the ground substance is an amorphous gel-like substance surrounding the cells. In a tissue, cells are surrounded and supported by an extracellular matrix. Ground substance traditionally does not include fibers (collagen and elastic fibers), but does include all the other components of the extracellular matrix . The components of the ground substance vary depending on the tissue. Ground substance is primarily composed of water, glycosaminoglycans (most notably hyaluronan ), proteoglycans, and glycoproteins. Usually it is not visible on slides, because it is lost during the preparation process. Connective Tissue Functions of Connective Tissues Support and connect other tissues Protection (fibrous capsules and bones that protect delicate organs and, of course, the skeletal system). Transport of fluid, nutrients, waste, and chemical messengers is ensured by specialized fluid connective tissues, such as blood and lymph. Adipose cells store surplus energy in the form of fat and contribute to the thermal insulation of the body. Embryonic Connective Tissue All connective tissues derive from the mesodermal layer of the embryo . The first connective tissue to develop in the embryo is mesenchyme , the stem cell line from which all connective tissues are later derived. -

Elastic Fibers: Building Bridges Between Cells and Their Matrix
Current Biology, Vol. 12, R279–R281, April 16, 2002, ©2002 Elsevier Science Ltd. All rights reserved. PII S0960-9822(02)00800-X Elastic Fibers: Building Bridges Dispatch Between Cells and Their Matrix Kim S. Midwood and Jean E. Schwarzbauer Other proteins, including the emerging family of fibulin proteins, contact elastic fibers in vivo and are thought to promote the formation and stabilization of Extracellular elastic fibers confer resilience and the fiber. Fibulin is derived from the Latin for clasp or flexibility to tissues. Recent studies have identified a buckle and there are currently five members of this protein, fibulin-5, that connects these fibers to cells family. The fibulins have overlapping but distinct pat- and regulates their assembly and organization. terns of expression and are particularly prominent in tissues rich in elastic fibers such as lung and blood vessels. Recent studies [5,6] have identified fibulin-5 In animals, cells within tissues specifically contact as a protein that links elastic fibers to cells and other cells. They also contact a complex network of regulates fiber assembly and organization. These secreted proteins and carbohydrates, the extracellu- complementary studies focus on the function of lar matrix. Animals contain many different types of fibulin-5 — also known as DANCE and EVEC — a extracellular matrix, each specialized for a different 66 kDa protein that co-localizes with, and binds to, function. For example, tendons exhibit great strength elastin on the surface of elastic fibers. Fibulin-5 also and the extracellular matrix in the kidney is designed binds to cells by interacting with integrin cell surface for filtration. -

Connective Tissues (C.T.)
Lecture 3: Connective tissues (C.T.) - Colours index : Red : important Grey : doctors notes Pink : Girls slides Objectives : 1. Enumerate the general characteristics of C.T. 2. Classify C.T. Into C.T. Proper (C.T.P.) and special types of C.T. 3. Describe components of C.T.P. 4. Classify C.T.P. and know the distribution and function of each type Definition and components of C.T. 1.It is one of the 4 basic tissues. 2.it is Mesodermal* in origin. Function of C.T 1. Supports, binds and connects other tissue and organs. 2. Provides structural (fix organ position) and metabolic support. General characteristics of C.T : 1. It is formed of widely separated, few cells with abundant extracellular matrix. 2. Most of C.T. Are vascular (have blood vessel). Components of C.T : 1. Cells: different types. 2. Fibers: collagenous, elastic & reticular. 3. Matrix: the intercellular substance = extracellular matrix, where cells and fibers are embedded. *Mesodermal: (the middle layer of an embryo in early development, between endoderm and ectoderm) “Referring to embryology” ;) Types of C.T. (Depending on matrix) - Soft = C.T. Proper - Rigid (firm,rubbery) = Cartilage - Hard (solid) = Bone - Fluid = Blood Components of C.T. Proper ● Cells ● Fibers ● Matrix Cells: 1. Fibroblasts 2. Macrophages 3. Mast cells 4. Plasma cells 5. Adipose cells 6. Leucocytes (اﻟﺧﻼﯾﺎ اﻟﻣﻛوﻧﺔ ﻟﻠـCells: (connective tissue ❖ Fibroblast Macrophages Mast Cells ● It’s the most common cell, L/M: L/M: found nearly in all types of C.T ● Basophilic cytoplasm, rich in Cytoplasm contains numerous proper. lysosomes. basophilic and cytoplasmic granules. -

Review: Epithelial Tissue
Review: Epithelial Tissue • “There are 2 basic kinds of epithelial tissues.” What could that mean? * simple vs. stratified * absorptive vs. protective * glands vs. other • You are looking at epithelial cells from the intestine. What do you expect to see? tight junctions; simple columnar; gobet cells; microvilli • You are looking at epithelial cells from the trachea. What do you expect to see? cilia; pseudostratified columnar; goblet cells 1 4-1 Four Types of Tissue Tissue Type Role(s) - Covers surfaces/passages - Forms glands - Structural support CONNECTIVE - Fills internal spaces - Transports materials - Contraction! - Transmits information (electrically) 2 Classification of connective tissue 1. Connective tissue proper 1a. Loose: areolar, adipose, reticular 1b. Dense: dense regular, dense irregular, elastic 2. Fluid connective tissue 2a. Blood: red blood cells, white blood cells, platelets 2b. Lymph 3. Supporting connective tissue 3a. Cartilage: hyaline, elastic, fibrocartilage 3b. Bone 3 Defining connective tissue by the process of elimination if not epithelial, muscle, or nervous, must be connective! 4 LAB MANUAL Figure 6.4 Areolar connective tissue: A prototype (model) connective tissue. Cell types Extracellular matrix Ground substance Macrophage Fibers = proteins • Collagen fiber • Elastic fiber • Reticular fiber Fibroblast Lymphocyte Adipocyte Capillary Mast cell 5 The Cells of Connective Tissue Proper Melanocytes and macrophages, mesenchymal, mast; Adipo- / lympho- / fibrocytes and also fibroblasts. These are the cells of connective -

Índice De Denominacións Españolas
VOCABULARIO Índice de denominacións españolas 255 VOCABULARIO 256 VOCABULARIO agente tensioactivo pulmonar, 2441 A agranulocito, 32 abaxial, 3 agujero aórtico, 1317 abertura pupilar, 6 agujero de la vena cava, 1178 abierto de atrás, 4 agujero dental inferior, 1179 abierto de delante, 5 agujero magno, 1182 ablación, 1717 agujero mandibular, 1179 abomaso, 7 agujero mentoniano, 1180 acetábulo, 10 agujero obturado, 1181 ácido biliar, 11 agujero occipital, 1182 ácido desoxirribonucleico, 12 agujero oval, 1183 ácido desoxirribonucleico agujero sacro, 1184 nucleosómico, 28 agujero vertebral, 1185 ácido nucleico, 13 aire, 1560 ácido ribonucleico, 14 ala, 1 ácido ribonucleico mensajero, 167 ala de la nariz, 2 ácido ribonucleico ribosómico, 168 alantoamnios, 33 acino hepático, 15 alantoides, 34 acorne, 16 albardado, 35 acostarse, 850 albugínea, 2574 acromático, 17 aldosterona, 36 acromatina, 18 almohadilla, 38 acromion, 19 almohadilla carpiana, 39 acrosoma, 20 almohadilla córnea, 40 ACTH, 1335 almohadilla dental, 41 actina, 21 almohadilla dentaria, 41 actina F, 22 almohadilla digital, 42 actina G, 23 almohadilla metacarpiana, 43 actitud, 24 almohadilla metatarsiana, 44 acueducto cerebral, 25 almohadilla tarsiana, 45 acueducto de Silvio, 25 alocórtex, 46 acueducto mesencefálico, 25 alto de cola, 2260 adamantoblasto, 59 altura a la punta de la espalda, 56 adenohipófisis, 26 altura anterior de la espalda, 56 ADH, 1336 altura del esternón, 47 adipocito, 27 altura del pecho, 48 ADN, 12 altura del tórax, 48 ADN nucleosómico, 28 alunarado, 49 ADNn, 28 -

Elastic Fiber Formation: a Dynamic View of Extracellular Matrix Assembly Using Timer Reporters
JOURNAL OF CELLULAR PHYSIOLOGY 207:87–96 (2006) Elastic Fiber Formation: A Dynamic View of Extracellular Matrix Assembly Using Timer Reporters BETH A. KOZEL,1 BRENDA J. RONGISH,2 ANDRAS CZIROK,2 JULIA ZACH,2 CHARLES D. LITTLE,2 ELAINE C. DAVIS,3 RUSSELL H. KNUTSEN,1 JESSICA E. WAGENSEIL,1 1 1 MARILYN A. LEVY, AND ROBERT P. MECHAM * 1Department of Cell Biology and Physiology, Washington University School of Medicine, St. Louis, Missouri 2Department of Anatomy and Cell Biology, University of Kansas Medical Center, Kansas City, Kansas 3Department of Anatomy & Cell Biology, McGill University, Montreal, Quebec, Canada To study the dynamics of elastic fiber assembly, mammalian cells were transfected with a cDNA construct encoding bovine tropoelastin in frame with the Timer reporter. Timer is a derivative of the DsRed fluorescent protein that changes from green to red over time and, hence, can be used to distinguish new from old elastin. Using dynamic imaging microscopy, we found that the first step in elastic fiber formation is the appearance of small cell surface-associated elastin globules that increased in size with time (microassembly). The elastin globules are eventually transferred to pre-existing elastic fibers in the extracellular matrix where they coalesce into larger structures (macroassembly). Mechanical forces associated with cell movement help shape the forming, extracellular elastic fiber network. Time-lapse imaging combined with the use of Timer constructs provides unique tools for studying the temporal and spatial aspects of extracellular matrix formation by live cells. J. Cell. Physiol. 207: 87–96, 2006. ß 2005 Wiley-Liss, Inc. Our understanding of the extracellular matrix (ECM) e-amino groups by a member of the lysyl oxidase enzyme has expanded greatly over the past two decades. -

The Role of Elastic Fibers in Pathogenesis of Conjunctivochalasis
Int J Ophthalmol, Vol. 10, No. 9, Sep.18, 2017 www.ijo.cn Tel:8629-82245172 8629-82210956 Email:[email protected] ·Review· The role of elastic fibers in pathogenesis of conjunctivochalasis Jing-Yun Gan, Qing-Song Li, Zhen-Yong Zhang, Wei Zhang, Xing-Ru Zhang Department of Ophthalmology, Putuo Hospital, Shanghai and even the whole body. Under the normal physiological University of Traditional Chinese Medicine, Shanghai 200062, conditions, elastic fibers are important components of China extracellular matrix, and influence the tissue flexibility and Correspondence to: Xing-Ru Zhang. Department of elasticity. Ophthalmology, Putuo Hospital, Shanghai University of In 1998, Meller and Tseng[2] proposed the hypothesis which Traditional Chinese Medicine, Shanghai 200062, China. states that mechanism of CCh is via accumulation of degrading [email protected] enzymes which resulted in elastotic degeneration and Received: 2016-11-22 Accepted: 2017-03-23 collagenolysis of bulbar conjunctiva in the tears as a result of delayed tear clearance. Abstract Although, a lot of research has been done on it, the precise ● The PubMed, MEDLINE databases and China National etiology of CCh remains obscure. So we summarize the Knowledge Infrastructure (CNKI) were searched for previous literatures and analyze the current situation on the information regarding the etiology and pathogenesis hypothesis of elastic fibers. of conjunctivochalasis (CCh) and the synthesis and MATERIALS AND METHODS degradation of elastic fibers. After analysis of the literature, The following electronic databases were screened: PubMed, we found elastic fibers was a complex protein molecule China National Knowledge Infrastructure (CNKI). The from the structure and composition; the degradation of following search equation was used: “CCh (all fields)” OR elastic fibers was one of the histopathological features “elastic tissue (MeSH terms)”, “elastic tissue (MeSH terms)” of the disease; the vast majority of the factors related to AND “2011/1/1 (PDAT):2016/5/31 (PDAT)”. -

Dysregulated Assembly of Elastic Fibers in Fibulin-5 Knockout Mice Results in a Tendon-Specific Increase in Elastic Modulus
journal of the mechanical behavior of biomedical materials 113 (2021) 104134 Contents lists available at ScienceDirect Journal of the Mechanical Behavior of Biomedical Materials journal homepage: http://www.elsevier.com/locate/jmbbm Dysregulated assembly of elastic fibers in fibulin-5 knockout mice results in a tendon-specific increase in elastic modulus Jeremy D. Eekhoff a, Heiko Steenbock b, Ian M. Berke a, Jürgen Brinckmann b,c, Hiromi Yanagisawa d, Jessica E. Wagenseil e, Spencer P. Lake a,e,f,* a Department of Biomedical Engineering, Washington University in St. Louis, USA b Institute of Virology and Cell Biology, University of Lübeck, Germany c Department of Dermatology, University of Lübeck, Germany d Life Science Center for Survival Dynamics, Tsukuba Advanced Research Alliance, University of Tsukuba, Japan e Department of Mechanical Engineering and Materials Science, Washington University in St. Louis, USA f Department of Orthopaedic Surgery, Washington University in St. Louis, USA ARTICLE INFO ABSTRACT Keywords: Elastic fiber assembly is coordinated in part by fibulin-5, a matricellular protein. When fibulin-5 is not available Fibulin-5 to guide elastogenesis, elastin forms into disconnected globules instead of the dense elastic fiber core found in Elastic fiber healthy tissues. Despite the growing evidence for a significant role of elastic fibers in tendon mechanics and the Tendon clinical relevance to cutis laxa, a human disease which can be caused by a mutation in the gene encoding fibulin- Orthopaedic Musculoskeletal 5, it is unknown how malformed elastic fibers affect tendon function. Therefore, this study investigated the Biomechanics effects of dysregulated elastic fiber assembly in tendons from fibulin-5 knockout mice in comparison to wild-type controls. -

Morfologia E Histoquímica Das Glândulas Sexuais Acessórias De Metachirus Nudicaudatus, Didelphidae-Marsupialia 883
Pesq. Vet. Bras. 36(9):881-892, setembro 2016 DOI: 10.1590/S0100-736X2016000900015 Morfologia e histoquímica das glândulas sexuais acessórias 1 de Metachirus nudicaudatus, Didelphidae-Marsupialia Suely F. Costa2*, José C. Nogueira3, Márcio G. Zangeronimo2, Bruno A. Soares4, Amália S. Chaves5 e Leandra Q. de Melo2 ABSTRACT.- Costa S.F., Nogueira J.C., Zangeronimo M.G., Soares B.A., Chaves A.S. & Melo L.Q. 2016. [Morphology and histochemistry of accessory sex glands of Metachirus nudicau- datus, Didelphidae-Marsupialia.] Morfologia e histoquímica das glândulas sexuais aces- sórias de Metachirus nudicaudatus, Didelphidae-Marsupialia. Pesquisa Veterinária Brasilei- ra 36(9):881-892. Departamento de Medicina Veterinária, Universidade Federal de Lavras, Campus Universitário, Lavras, MG 37200-000, Brazil. E-mail: This paper describes the morphology and distribution of glycogen and mucous subs- tances in the prostate and the bulbourethral glands of [email protected] nudicaudatus (Geoffroy, 1803), the only species of the genus. The prostate is surrounded by the tunica adventitia, and muscle and stroma is formed by connective urethral mucosa. The glandular parenchy- ma consists of secretory tubules, scattered throughout the connective tissue of the ure- thral mucosa which differs histologically and histochemically in cranial, middle, and caudal segments of the prostate. These morpho-histochemical differences are also observed in the outer, middle and inner parts of the tubular epithelium of each prostatic segment. In general, prostatic segments secrete neutral mucous substances, and the caudal segment also produces glycogen. The three pairs of bulbourethral glands (lateral, intermediate and medial) are surrounded by a capsule of dense connective tissue and skeletal striated mus- cle.